绪论
疾病是一个极其复杂的过程。在病原因子和机体反应功能的相互作用下,患病机体有关部分的形态结构、代谢和功能都会发生种种改变,这是研究和认识疾病的重要依据。病理学(patho1ogy)的任务就是运用各种方法研究疾病的原因(病因学,ethiology)、在病因作用下疾病发生发展的过程(发病学,pathogenesis)以及机体在疾病过程中的功能、代谢和形态结构的改变(病变,patho1ogica1 changes),阐明其本质,从而为认识和掌握疾病发生发展的规律,为防治疾病,提供必要的理论基础。
一、病理学的内容和任务
本书第1~7章属病理学总论内容,第8~19章属各论内容。前者研究各种疾病的共同的基本规律,后者则研究各个器官或器官系统疾病的特殊规律。
各个器官虽然在功能和结构上互不相同,但在各种致病因子的影响下,不同器官却可呈现同样的基本反应和结构改变,这就是病理学总论的研究对象和内容。例如,肝炎、肺炎、脑膜炎、阑尾炎、腹膜炎等,虽然各有其本身的病因和独特的病变,并发生于不同的器官,但却都属炎性疾患,都具有细胞、组织损伤,局部血液循环障碍,炎性渗出和细胞、组织增生等共同的炎症的基本改变,其本质也都是病因对机体的损伤和机体对损伤的防御反应在相应局部的表现。病理学总论是阐述细胞和组织的损伤、损伤的修复、局部血液循环及体液循环障碍、免疫病理、炎症、遗传与疾病以及肿瘤等基本病理过程及其发生发展的基本规律,阐明其本质,以便运用这些知识去更深刻地发现和认识各种疾病的特殊规律和本质。
然而,各个疾病又各有自身的病因、发病机制、好发部位及其形态学改变和相应的临床表现。病理学各论就是阐明各种疾病的病因、病变及其发生发展的特殊规律,研究其与临床表现的关系及其对疾病防治的意义。
显然,病理学总论与各论之间有着密切的内在联系,学好总论是学习各论的必要基础,学习各论也必须联系运用总论知识,同时加深对总论的理解,两者互相联系,密切相关,学习时不可偏废。
二、病理学在医学中的地位
随着自然科学的发展,医学科学逐渐形成了许多分支学科,它们的共同目的和任务就是从不同角度、用不同方法去研究正常和患病机体的生命活动,为防治疾病,保障人类健康服务。病理学除侧重从形态学角度研究疾病外,也研究疾病的病因学、发病学以及形态改变与功能变化及临床表现的关系。因此,病理学与基础医学中的解剖学、组织学、胚胎学、生理学、生物化学、寄生虫学、微生物学等均有密切的联系,也是学习临床医学的重要基础,是基础医学与临床医学之间的桥梁。
病理学与临床医学之间的密切联系,明显地表现在对疾病的研究和诊断上。临床医学除运用各种临床诊察、检验、治疗等方法对疾病进行诊治外,往往还必须借助于病理学的研究方法如活体组织检查、尸体剖检以及动物实验等来对疾病进行观察研究,提高临床工作的水平。病理学则除进行实验研究(实验病理学)外,也必须密切联系临床,直接从患病机体去研究疾病,否则也不利于病理学本身的发展。
三、病理学的研究方法
病理学的研究方法多种多样,研究材料主要来自自患病人体(人体病理材料)和实验动物以及其他实验材料如组织培养、细胞培养等(实验病理材料)。
(一)尸体剖检
对死亡者的遗体进行病理剖检(尸检)是病理学的基本研究方法之一。尸体剖检(autopsy)不仅可以直接观察疾病的病理改变,从而明确对疾病的诊断,查明死亡原因,帮助临床探讨、验证诊断和治疗是否正确、恰当,以总结经验,提高临床工作的质量,而且还能及时发现和确诊某些传染病、地方病、流行病、为防治措施提供依据,同时还可通过大量尸检积累常见病、多发病、以及其他疾病的人体病理材料,为研究这些疾病的病理和防治措施,为发展病理学作贡献。显然,尸检是研究疾病的极其重要的方法和手段,人体病理材料则是研究疾病的最为宝贵的材料。
一个国家尸检率的高低往往可以反映其文明进步的程度,世界上不少文明先进国家的尸检率达到90%以上,有的国家在法律中对尸检作了明文规定。我国的尸检率还很低,十分不利于我国病理学和医学科学的发展,亟待提高。
(二)活体组织检查
用局部切除、钳取、穿刺针吸以及搔刮、摘除等手术方法,由患者活体采取病变组织进行病理检查,以确定诊断,称为活体组织检查(biopsy),简称活检。这是被广泛采用的检查诊断方法。这种方法的优点在于组织新鲜,能基本保持病变的真像,有利于进行组织学、组织化学、细胞化学及超微结构和组织培养等研究。对临床工作而言,这种检查方法有助于及时准确地对疾病作出诊断和进行疗效判断。特别是对于诸如性质不明的肿瘤等疾患,准确而及时的诊断,对治疗和预后都具有十分重要的意义。
(三)动物实验
运用动物实验的方法,可以在适宜动物身上复制某些人类疾病的模型,以便研究者可以根据需要,对之进行任何方式的观察研究,例如可以分阶段地进行连续取材检查,以了解该疾病或某一病理过程的发生发展经过等。此外,还可利用动物实验研究某些疾病的病因、发病机制以及药物或其他因素对疾病的疗效和影响等。这种方法的优点是可以弥补人体观察之受限和不足,但动物与人体之间毕竟存在种种差异,不能将动物实验的结果直接套用于人体,这是必须注意的。
(四)组织培养与细胞培养
将某种组织或单细胞用适宜的培养基在体外加以培养,以观察细胞、组织病变的发生发展、如肿瘤的生长、细胞的癌变、病毒的复制、染色体的变异等等。此外,也可以对其施加诸如射线、药物等外来因子,以观察其对细胞、组织的影响等。这种方法的优点是,可以较方便地在体外观察研究各种疾病或病变过程,研究加以影响的方法,而且周期短、见效快,可以节省研究时间,是很好的研究方法之一。但缺点是孤立的体外环境毕竟与各部分间互相联系、互相影响的体内的整体环境不同,故不能将研究结果与体内过程等同看待。
(五)病理学的观察方法
近年来,随着学科的发展,病理学的研究手段已远远超越了传统的经典的形态观察,而采用了许多新方法、新技术,从而使研究工作得到了进一步的深化,但形态学方法(包括改进了的形态学方法)仍不失为基本的研究方法。兹将常用的方法简述如下:
大体观察 主要运用肉眼或辅之以放大镜、量尺、各种衡器等辅助工具,对检材及其病变性状(大小、形态、色泽、重量、表面及切面状态、病灶特征及坚度等)进行细致的观察和检测。这种方法简便易行,有经验的病理及临床工作者往往能借大体观察而确定或大致确定诊断或病变性质(如肿瘤的良恶性等)。
组织学观察将病变组织制成厚约数微米的切片,经不同方法染色后用显微镜观察其细微病变,从而千百倍地提高了肉眼观察的分辨能力,加深了对疾病和病变的认识,是最常用的观察、研究疾病的手段之一。同时,由于各种疾病和病变往往本身具有一定程度的组织形态特征,故常可借助组织学观察来诊断疾病,如上述的活检。
细胞学观察运用采集器采集病变部位脱落的细胞,或用空针穿刺吸取病变部位的组织、细胞,或由体腔积液中分离所含病变细胞,制成细胞学涂片,作显微镜检查,了解其病变特征。此法常用于某些肿瘤(如肺癌、子宫颈癌、乳腺癌等)和其他疾病的早期诊断。但限于取材的局限性和准确性,有时使诊断难免受到一定的限制。既提高了穿刺的安全性,也提高了诊断的准确性。
超微结构观察运用透射及扫描电子显微镜对组织、细胞及一些病原因子的内部和表面超微结构进行更细微的观察(电子显微镜较光学显微镜的分辨能力高千倍以上),即从亚细胞(细胞器)或大分子水平上认识和了解细胞的病变。这是迄今最细致的形态学观察方法。在超微结构水平上,还常能将形态结构的改变与机能代谢的变化联系起来,大大有利于加深对疾病和病变的认识。
组织化学和细胞化学观察通过运用具有某种特异性的、能反映组织和细胞成分化学特性的组织化学和细胞化学方法,可以了解组织、细胞内各种蛋白质、酶类、核酸、糖原等等化学成分的状况,从而加深对形态结构改变的认识。这种方法不仅可以揭示普通形态学方法所不能观察到的组织、细胞的化学成分的变化,而且往往在尚未出现形态结构改变之前,就能查出其化学成分的变化。此外,随着免疫学技术的进步,还可运用免疫组织化学和免疫细胞化学的方法,了解组织、细胞的免疫学性状,对于病理学研究和诊断都有很大帮助。
除上述常用方法外,近数十年来陆续建立的还有放射自显影技术、显微分光技术、形态测量(图像分析)技术、分析电镜技术、流式细胞仪(FCM)技术、多聚酶链反应(PCR)技术以及分子原位杂交技术等一系列分子生物学技术,从而使常规的病理形态学观察,发展到将形态结构改变与组织、细胞的的化学变化结合志来进行研究,而且将历来的定性的研究发展到对病理改变进行形态的和化学成分的定量研究,从而获得了大量的更多更新的新信息,大大加深了疾病研究的深度。这是以往的研究所难以实现的。
四、病理学的发展
病理学是在人类探索和认识自身疾病的过程中应运而生的。它的发展自必受到人类认识自然能力的制约。从古希腊的Hippocrates开始,经过2千多年的发展,直到18世纪中叶,由于自然科学的兴起,促进了医学的进步,意大利医学家Morgagni(1682-1771)根据积累的尸检材料创立了器官病理学(organ pathology), 标志着病理形态的开端。约一个世纪以后的19世纪中叶,德国病理学家Virchow(1821~1902)在显微镜的帮助下,首创了细胞病理学(celluar pathology),不仅对病理学而且对整个医学的发展作出了具有历史意义的、划时代的贡献。直到今天,他的学说还继续影响着现代医学的理论和实践。
我国秦汉时期的《黄帝内经》、隋唐时代巢元方的《诸病源候论》、南宋时期宋慈的《洗冤集录》等世界名著,对病理学的发展作出了很大的贡献。半个多世纪以来,我国现代病理学先驱徐育明、胡正详、梁伯强、谷镜汧、侯宝璋和林振纲、秦光煜、江晴芬、李佩琳、吴在东、杨述祖、杨简、刘永等为我国病理学教学、师资培养以及病理学的发展,呕心沥血,艰辛创业,功绩卓著。在他们的主持和参与下,我国从无到有地编著了自己的具有我国特色的病理学教科书和参考书。同时,大力推进我国的病理尸检和活检工作以及科研工作,对长期以来严重危害我国人民健康的地方病和寄生虫病(如克山病、大骨节病、黑热病、血吸虫病等)、肿瘤(如肝癌、食管癌、鼻咽癌等)以及心血管疾病(如动脉粥样硬化症、冠心病等)等常见病、多发病、进行了广泛深入的研究,到得了丰硕的成果。这些成就不仅对我国当前病理学教学、科研和检验工作,而且对今后我国病理的发展,都起着重要的作用。
病理的发展与自然科学,特别是基础科学的发展和技术进步有着密切的联系。当人们还只能依赖肉眼和简单的放大镜观察事件时,便只能产生器官病理学;只有到了显微镜和细胞学问世之后,才有可能诞生了细胞病理学;而半个多世纪以来,由于电子显微镜技术、特别是20多年来一系列有关新方法新技术的相继建立和细胞生物学、分子生物学、环境医学以及现代免疫学、现代遗传学等新兴学科及其分支的迅速兴起和发展,对医学科学、也对病理学的发展产生了深刻的影响,带来了新的动力。近年来,超微病理学(ultra-structural pathology)包括分子病理学(molecular pathology),以及分子免疫学(molecular immunology)、分子遗传学(molecular genetics)等等新的边缘学科和学科分支的建立,促使病理学已不仅从细胞和亚细胞水平,而且深入到从分子水平、从人类遗传基因突变和染色体畸变等去认识有关疾病,研究疾病的起因和发病机制。现代遗传病理学认为,在人类疾病中虽然只有一小部分具有明显的遗传特征,但原则上几乎所有疾病都受遗传因素的影响。现代免疫病理学的研究则逐步阐明了许多长期以来未被认识的疾病的病因、发病机制及其本质,发现许多疾病的发生发展均与机体的免疫状态密切相关。这些进展和发现,为许多疾病的防治开辟了新的前景。
我国病理在前辈病理学者奠定的坚实基础上,经过新一代病理学者的努力,已有了长足的进步,在队伍和条件的建设上得到了显著的发展。我国是一个幅员辽阔、人口众多的大国,疾病谱和疾病都具有自己的特点,开展好这方面的研究,不仅对我国医学发展和疾病防治具有极为重要的意义,同时也是对世界医学的贡献。面对这一任务,我国病理学的发展具有充分的现实条件和广阔的前景。当前,我们既要面对现实,大力提倡和开展病理尸检工作,充分利用我国充丰富的疾病材料“资源”,积极发展我国的人体病理学,也要充分利用各种途径吸收世界上的新方法新技术,同时这要根据我国的实际情况,开发和建立自己的新方法新技术,加强我国的实验病理学研究,使我国病理学的发展跟上世界病理学发展的步伐,并在某些方面处于领先地位。这当是我国当代病理学者的责任和任务。
第一章 细胞和组织的损伤
第一节 概述
生活机体的细胞和组织经常不断地接受内外环境各种刺激因子的影响,并通过自身的反应和调节机制对刺激作出应答反应。这种反应能力可保证细胞和组织的正常功能,维护细胞、器官乃至整个机体的生存。但细胞和组织并非能适应所有刺激的影响,当刺激的性质、强度和持续时间超越了一定的界限时,细胞乃受损甚至死亡。
然而,在正常和发生了适应性改变的、损伤的死亡的细胞之间,在结构和功能上往往并无截然的界限。可复性的和不可复性的改变之间,也常难以截然区分。总之,这些过程都是逐渐过渡的。至于一个刺激究竟会引行细胞的适应性改变、损伤还是死亡,也只有待其作用一定的时间(潜伏期),细胞和组织出现明确的结构变化以后,才能从形态上加以判断。这段潜伏期的长短不仅决定于刺激因子的性质和强度,还取决于受累细胞和组织的种类。例如中枢神经系统特别是神经节细胞对于缺氧的耐受能力就远不如结缔组织细胞,也不如肝、肾、肺等器官的实质细胞。常温下大脑缺氧后尚能复苏的时间极限为8~10分钟(大脑壳核又比其他部位如脑干核团更为敏感),肝通常为30~35分钟,肾为60~180分钟,肺约为60分钟(支气管上皮约为90分钟)。
当然对于细胞损伤的观察又和所用方法的灵敏度和分辨能力有关。例如在肝缺血时,5分钟后即可从生化上证明其氧化磷酸化过程明显降低,而最初的结构改变如内质网、线粒体、细胞膜等亚细胞结构的改变,则约在缺血15分钟后才能在电子显微镜下检见,但这些改变通常还是可复性的,只要在30分钟内能恢复血流灌注,则这些改变仍可恢复正常。然而此时在光学显微镜下,一般尚不见明显的改变。只有当缺血持续达30分钟以上时,细胞的损伤才达到不可复的程度,并趋于死亡。但也要在缺血达2小时以后才能在光学显微镜下检见肯定的细胞损伤。当然,肉眼观能检见的改变还远远在此以后。
第二节 细胞的超微结构及其基本病理过程
Virchow在19世纪中期所奠定的细胞病理学说,通过近代对细胞及其病变的超微结构以及结构与功能相结合的研究,已经获得了新的更广更深的基础,扩大和加深了对疾病的理解。
细胞是一个由细胞膜封闭的基本生命单元,内含一系列明确无误的互相分隔的反应腔室,这就是由细胞膜为界限的各种细胞器,是细胞代谢和细胞活力的形态支柱。细胞内的这种严格分隔保证各种细胞器分别进行着无数的生化反应,行使各自的独特功能,维持细胞和机体的生命活动。细胞器的改变是各种病变的基本组成部分。
一、细胞核
细胞核(nucleus)是遗传信息的载体,细胞的调节中心,其形态随细胞所处的周期阶段而异,通常以间期核为准。
细胞核外被核膜。核膜由内外二层各厚约3nm的单位膜构成,中间为2~5nm宽的间隙(核周隙);核膜上有直径约50nm的微孔,作为核浆与胞浆间交通的孔道,其数目因细胞类型和功能而异,多者可占全核表面积的25%;在肝细胞核据估算约有2000个核孔。
核浆主由染色质构成,其主要成分为脱氧核糖核酸(DNA),并以与蛋白质相结合的形式存在,后者由组蛋白与非组蛋白组成。染色质的NDA现在已可用多种方法加以鉴定和定量测定。
核内较粗大浓缩的、碱性染料深染的团块状染色质为异染色质,呈细颗粒状弥散分布的、用普通染色法几乎不着色的染色质则为常染色质。一部分异染色质也可以上述两种状态存在。从生化角度看,异染色质不具遗传活性,相反,常染色质则大部分具遗传活性。
间期核的染色质模式还反映细胞的功能状态。一般而言,大而淡染的核(浓缩染色质少)提示细胞活性(如蛋白质和酶的合成)较高;小而深染的核(浓缩染色质较多)则提示细胞活性有限或降低。
(一)细胞损伤时核的改变
1、核大小的改变核的大小通常反映着核的功能活性状态,功能旺盛时核增大,核浆淡染,核仁也相应增大和(或)增多。如果这种状态持续较久,则可出现多倍体核或形成多核巨细胞。多倍体核在正常情况下亦可见于某些功能旺盛的细胞,如肝细胞中可见约20%为多倍体核。在病理状态下,如晚期肝炎及实验性肝癌前期等均可见多倍体的肝细胞明显增多。
核的增大除见于功能旺盛外,也可见于细胞受损时,最常见的情况为细胞水肿。这主要是细胞量匮乏或毒性损伤所致,是核膜钠泵衰竭导致水和电解质运输障碍的结果。这种核肿大又称为变性性核肿大。
相反,当细胞功能下降或细胞受损时,核的体积则变小,染色质变致密,如见于器官萎缩时。与此同时核仁也缩小。
2.核形的改变光学显微镜下,各种细胞大多具有各自形状独特的核,可为圆形、椭圆形、梭形、杆形、肾形、印戒形、空洞形以及奇形怪状的不规则形(图1-1)等。在电镜下由于切片极薄,切面可以多种多样,但均非核的全貌。核的多形性和深染特别多见于恶生肿瘤细胞,称为核的异型性(atypia)。
3.核结构的改变细胞在衰亡及损伤过程中的重要表征之一是核的改变,主要表现为核膜和染色质的改变。
核浓缩(karyopyknosis):染色质在核浆内聚集成致密浓染的大小不等的团块状,继而整个细胞核收缩变小,最后仅留下一致密的团块,是为核浓缩。这种浓缩的核最后还可再崩解为若干碎片(继发性核碎裂)而逐渐消失。
核碎裂(karyorrhexis):染色质逐渐边集于核膜内层,形成较大的高电子密度的染色质团块(图1-2)。核膜起初尚保持完整,以后乃在多处发生断裂,核逐渐变小,最后裂解为若干致密浓染的碎片。
核溶解(karyolysis):变致密的结成块状的染色质最后完全溶解消失。即核溶解。核溶解变可不经过核浓缩或核碎裂而一开始即独立进行。在这种情况下,受损的核很早就消失。
上述染色质边集(即光学显微镜下所谓的核膜浓染)、核浓缩、核碎裂、核溶解等核的结构改变为核和细胞不可复性损伤的标志,提示活体内细胞死亡(坏死)。
4.核内包含物(intranuclear inclusions) 在某些细胞损伤时可见核内出现各种不同的包含物,可为胞浆成分(线粒体、内质网断片、溶酶体、糖原颗粒、脂滴等),亦可为非细胞本身的异物,但最常见的还是前者。这种胞浆性包含物可在两种情况下出现:①胞浆成分隔着核膜向核内膨突,以致在一定的切面上看来,似乎胞浆成分已进入核内,但实际上大多仍可见其周围有核膜包绕,其中的胞浆成分常呈变性性改变(如髓鞘样结构,膜碎裂等)。这种包含物称为胞浆性假包含物(图1-3);②在有丝分裂末期,某些胞浆结构被封入形成中的子细胞核内,以后出现于子细胞核中,称为真性胞浆性包含物。
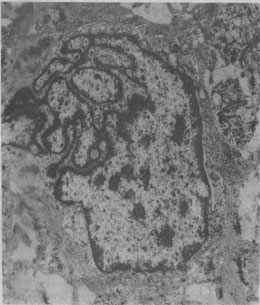
图1-1 恶性肿瘤细胞的奇异形核
图中央为一巨大的瘤细胞核,核膜曲折凹陷,使核呈奇异形(纤维肉瘤的电镜照片)
■[此处缺少一些内容]■
5.核仁的改变核仁(nucleolus)为核蛋白体RNA转录和转化的所在。除含蛋白的均质性基质外,电镜下核仁主由线团状或网状电子致密的核仁丝(nucleolonema)和网孔中无结构的低电子密度的无定形部(pars amorpha)组成。核仁无界膜,直接患浮于核浆内。
形态上和生物上核仁由3种不同的成分构成:①原纤维状成分,内含蛋白质及与其相结合的45S-rRNA;②细颗粒状成分,主要由12S-rRNA构成,为核仁的嗜碱性成分;③细丝状成分,仅由来自胞浆的蛋白质构成,穿插于整个核仁内。3种核仁成分的空间排列状态可反映细胞的蛋白合成活性,例如:
壳状核仁:原纤维状成分集中位于核仁中央,细颗状成分呈壳状包绕于外层。这种细胞的合成活性甚低。
海绵状核仁:这种核仁的原纤维状与细颗状成分呈海绵状(或线团状)排列。这种细胞的合成活性升高。大多数所谓的“工作核”具有这种核仁。
高颗粒性核仁:由海绵状核仁转化而成,原纤维状成分几乎消失,核仁主要由颗粒状成分构成,故组织学上呈强嗜碱性,细胞的合成活性旺盛。这种核仁常见于炎症和肿瘤细胞。
低颗粒性核仁,与上述高颗粒性核仁相反,这种核仁的细颗粒状成分锐减,故电镜下原纤维状成分显得突出,电子密度较低。这种核仁常见于再生时,因此时细颗粒成分(rRNA)过多地被胞浆所利用。
分离性核仁:超微结构上3种核仁成分清楚地互相分离,原纤维状和细颗粒状成分减少。这种核仁变小,无活性,常见于核仁转录过程被抗生素、细胞抑制剂、缺氧和蝇菌素中毒等所完全阻断时。
由此可见,核仁的大小和(或)数目的多少常反映细胞的功能活性状态:大和(或)多的核仁是细胞功能活性高的表现,反之则细胞功能活性低。
二、细 胞 膜
细胞膜是包于细胞表面、将细胞与周围环境隔开的弹性薄膜,厚约8~10nm,由脂质和蛋白质构成,故为脂蛋白膜,对于细胞的生命活动和功能具有十分重要的意义。
细胞膜在许多特定场合可向外形成大量的纤细突起(微绒毛、纤毛),或向内形成各种形式的内褶(图1-4),
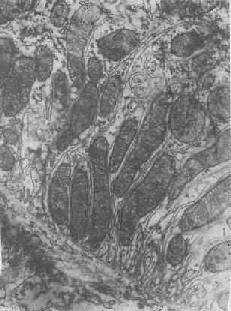
图1-4 肾近曲小管上皮细胞之基底褶及其中的线粒体
以利于其功能活动。相邻细胞的细胞膜之间还可形成闭锁小带、附着小带、桥粒和缝隙连接等各种特化结构,以保持细胞间的联系。此外,新近还发现,在相领细胞膜上有“粘附分子(如adhesion molecule,cadherin)”,对细胞正常结构和联系以及细胞极性的维持和细胞的分化等,均具有重要作用。
细胞膜除作为细胞的机械性和化学性屏障外,还具有一系列重要的功能诸如细胞内外的物质交换、细胞运动、细胞识别以及细胞的生长调控、免疫决定和各种表面受体形成等。
细胞的物质交换:细胞内外的物质交换主要以两种方式进行,一为渗透,一为出入胞过程。渗透乃指低分子物质(主要为水和电解质)通过细胞膜进出细胞,又可分两种情况:一种是按该物质在细胞内外环境中的浓度差,由浓高的一侧弥散底到低的一侧(被动运输);另一种则逆浓度差进行,即由浓度低的一侧向浓度高的一侧输送(主动运输),其经典的例子即Na+和K+的运输(在细胞内Na+和K+的比例为1:20,而在细胞外隙则为30:1),即依靠所谓“钠泵”的作用将Na+移向细胞外隙,而使K+移向细胞内。这种主动运输是一个耗能的过程,并由Na+和K+激活细胞膜上的ATP酶分解ATP而提供所需的能量。因此,如ATP酶受到某些毒物的抑制,则这种主动运输过程也同样受到阻抑。除Na+和K+外,其他一些有机物质如葡萄糖、氨基酸以及一些低分子代谢产物也是借这样的过程运输的。
第二种物质运输方式为出入胞过程。较大的分子和颗粒不能借渗透过程通过细胞膜,乃借出入胞过程将细胞内物质运送到细胞外和将细胞外物质移入细胞内。前者称为出胞(exocytosis),后者称为入胞(endocytosis)。进入细胞的如为液态物质则称之为胞饮或吞饮(pinocytosis),如为固体颗粒(如细菌、尘粒等异物)则称之为吞噬(phagocytosis)。在吞饮过程中,被吞饮的物质先接触并附着于细胞膜上,然后该处细胞膜连同该物质内凹,继而从细胞膜上断离下来,在胞浆内形成有膜包绕的小泡(吞饮小泡);吞噬过程与吞饮相似,稍不同的是,被吞噬物附着于细胞膜上后,细胞膜乃形成伪足样突起,将该物质环抱,最后封闭成有膜包绕的泡状结构,从而将该物质移入胞浆内(吞噬泡或吞噬体)。细胞自身的成分如蛋白质分子、糖原颗料、衰变的或受损而待处理的细胞器等,亦可被膜包绕而形成自噬泡(autophagic vacuoles)或自噬体(autophagosome)。胞饮泡或吞噬泡一般在胞浆内与溶酶体相结合,并被溶酶体酶所降解消化。但胞饮泡也可不经处理而穿过胞浆,最后从细胞的另一极重新移出细胞外。
细胞膜上还有特殊的识别区,结合在糖萼上,借此,细胞可相互识别,从而相互接近形成一定的细胞组合,或相互排斥而分离。同样,通过识别区,增生中的细胞在互相接触时就会停止分裂(接触抑制),而癌细胞则已失去这种表面功能,故可不受限制地增生。此外,细胞膜上还有一种膜抗原可以识别“自我”和“非我”(组织相容性抗原)。这种膜抗原在器官移植中具有重要意义,因为它可致敏受体,从而引起对移植物的排斥反应。最后,细胞上还有一些特异性区域带着特殊的化学簇,可以接受相应的化学信号,称为膜受体或表面受体。但从形态学上不能辨认。这种膜受体具有十分重要的意义,因为已知许多物质如激素、免疫球蛋白、药物、毒素以及感染因子等都是作为外来信号被受体接受后才转化为细胞内效应而发挥其作用。如封闭其受体,则亦同时消除其作用。
细胞膜的病变
1.细胞膜形态结构的改变 机械力的作用或细胞强烈变形,可引起红细胞膜的破损,如人工心瓣膜可引起细胞膜的破裂;某些脂溶性阴离子物质、溶蛋白和溶脂性酶以及毒素等也能破坏细胞膜的完整性。细胞膜结构的损伤可导致细胞内容物的外溢或水分进入细胞使细胞肿胀。
2.细胞膜通透性的改变 能量代谢不足(如缺氧时)或毒物的直接损害等所致各种不同的细胞损伤时,均可造成细胞主动运输的障碍,从而导致细胞内Na+的潴留和K+的排出,但Na+的潴留多于K+的排出,使细胞内渗透压升高,水分因而进入细胞,引起细胞水肿。这种单纯的通透性障碍时并不见细胞膜的形态学改变,只有借细胞化学方法才可在电镜下检见细胞膜上某些酶(如ATP酶、硷性磷酸酶、核苷酸酶等)活性的改变。当然,如损伤或水肿严重,则亦可发生继发性形态改变如出现肿浆膨出、微绒毛变短甚至消失、细胞膜基底变平乃至细胞膜破裂等。在某些较严重的损伤时还可出现细胞膜的螺旋状或同心圆层状卷曲,形成典型的髓鞘样结构(myelin figure)(图1-5)
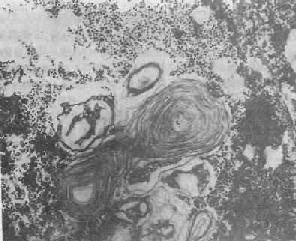
图1-5 髓鞘样结构
三、内质网
除红细胞外,内质网或多或少地见于所有各种细胞。内质网为由生物膜构成的互相通连的片层隙状或小管状系统,膜片间的隙状空间称为池,通常与细胞外隙和细胞浆基质之间不直接相通。这种细胞内的膜性管道系系一方面构成细胞内物质运输的通路,另方面为细胞内各种各样的酶反应提供广阔的反应面积。内质网与高尔基体及核膜相连续。
(一)粗面内质网
在病理状态下,粗面内质网(RER)可发生量和形态的改变。在蛋白质合成及分泌活性高的细胞(如浆细胞、胰腺腺泡细胞、肝细胞等)以及细胞再生和病毒感染时,粗面内质网增多。粗面肉质岗的含量高低也常反映肿瘤细胞的分化程度。相反,在萎缩的细胞(如饥饿时)以及有某种物质贮积的细胞,其粗面内质网则萎缩、减少。当细胞受损时,粗面内质网上的核蛋白体往往脱落于胞浆内,粗面内质网的蛋白合成乃下降或消失;当损伤恢复时,其蛋白合成也随之恢复。
在由各种原因引起的细胞变性和坏死过程中,粗面内质网的池一般出现扩张,较轻的和局限性的扩张只有在电镜下才能窥见,重度扩张时则在光学显微镜下可表现为空泡形成,电镜下有时可见其中含有中等电子密度的絮状物。在较强的扩张时,粗面内质网同时互相离散,膜上的颗粒呈不同程度的脱失。进而内质网本身可断裂成大小不等的片段和大小泡(图1-6)。这些改变大多见于细胞水肿时,故病变不仅见于内质网,也同时累及Golgi器、线粒体和胞浆基质,有时甚至还累及溶解体。
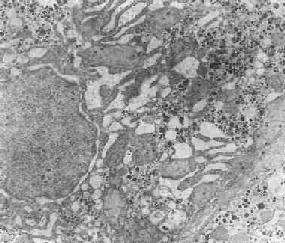
图1-6肝细胞粗面内质网扩张
(二)光面内质网
光面内质网的功能多种多样,即参与糖原的合成,又能合成磷脂、糖脂以及糖蛋白中的糖成分,此外,还在甾类化合物的合成中起重要的作用,故在合成甾类激素的细胞中特别丰富。光面内质网含有脱甲基酶、脱羧酶、脱氨酶、葡糖醛酸酶以及混合功能氧化酶等,因而光面内质网能分解甾体、能灭活药物和毒物并使其能被排除(如肝细胞)。肠上皮细胞的光面内质网参与脂肪的运输,心肌细胞的光面内质网(肌浆网)则参与心肌的刺激传导。
在生理状态下,随着细胞功能的升降,光面内质网(SER)的数量也呈现相应改变。但亦可出现完全相反的情况,例如在某些疾病(如淤胆)时,从形态结构上看,肝细胞光面内质网显著增生(图1-7),但其混合功能氧化酶的活性反而下降,这实际上是细胞衰竭的表现。
肝细胞的光面内质网具有生物转化作用(biotransformation),能对一些低分子物质如药物、毒品、毒物等,进行转化解毒,并将间接胆红素转化为直接胆红素。
许多成瘾药物和嗜好品如巴比妥类、吸毒、嗜酒等,可导致肝细胞光面内质网的增生,长期服用口服避孕药、安眠药、抗糖尿病药等也能导致同样后果。在HBsAg阳性肝炎时,肝细胞内光面内质网明显增生,在其管道内形成HBsAg。由于光面内质网的大量增生,这种肝细胞在光学显微镜下呈毛玻璃外观,故有毛玻璃细胞之称,并可为地衣红(orcein)着染。
在细胞损伤时光面内质网也可出现小管裂解为小泡或扩大为大泡状。在药物及某些芳香族化合物(主为致癌剂)的影响下,光面内质网有时可在胞浆内形成葱皮样层状结构,即“副核”,可为细胞的适应性反应(结构较松)或为变性性改变(结构致密)。
图1-7 肝细胞光面内质网增生伴轻度扩大
四、线粒体
线粒体(mitochondrion)是细胞内主要的能量形成所在,故不论在生理上或病理上都具有十分重要的意义。
线粒体为线状、长杆状、卵圆形或圆形小体,外被双层界膜。外界膜平滑,内界膜则折成长短不等的嵴并附有基粒。内外界膜之间为线粒体的外室,与嵴内隙相连,内界膜内侧为内室(基质室)(图1-8)。在合成甾类激素的内分泌细胞(如肾上腺皮质细胞、卵甾滤泡细胞、睾丸的Leydig细胞等),线粒体嵴呈小管状。内外界膜的通透性不同,外界膜的通透性高,可容许多种物质通过,而内界膜则构成明显的通透屏障,使一些物质如蔗糖和NADH全然不能通过,而其他物质如Na+ 和Ca 2+等也只有借助于主动运输才能通过。线粒体的基质含有电子致密的无结构颗粒(基质颗粒),与二价阳离子如Ca2+及Mg2+具有高度亲和力。基质中进行着β氧化、氧化脱羧、枸橼酸循环以及尿素循环等过程。在线粒体的外界膜内含有单胺氧化酶以及糖和脂质代谢的各种转移酶;在内界膜上则为呼吸链和氧化磷酸化的酶类。
线粒体是对各种损伤最为敏感的细胞器之一。在细胞损伤时最常见的病理改变可概括为线粒体数量、大小和结构的改变:
1.数量的改变线粒体的平均寿命约为10天。衰亡的线粒体可通过保留的线粒体直接分裂为二予以补充。在病理状态下,线粒体的增生实际上是对慢性非特异性细胞损伤的适应性反应或细胞功能升高的表现。例如心瓣膜病时的心肌线粒体、周围血液循环障碍伴间歇性跛行时的骨骼肌线粒体的呈增生现象。
线粒体数量减少则见于急性细胞损伤时线粒体崩解或自溶的情况下,持续约15分钟。慢性损伤时由于线粒体逐渐增生,故一般不见线粒体减少(甚至反而增多)。此外,线粒体的减少也是细胞未成熟和(或)去分化的表现。
2.大小改变细胞损伤时最常见的改变为线粒体肿大。根据线粒体的受累部位可分为基质型肿胀和嵴型肿胀二种类型,而以前者为常见。基质型肿胀时线粒体变大变圆,基质变浅、嵴变短变少甚至消失(图1-9)。在极度肿胀时,线粒体可转化为小空泡状结构(图1-10,图1-11)。此型肿胀为细胞水肿的部分改变。光学显微镜下所谓的浊肿细胞中所见的细颗粒即肿大的线粒体。嵴型肿较少见,此时的肿胀局限于嵴内隙,使扁平的嵴变成烧瓶状乃至空泡状,而基质则更显得致密。嵴型肿胀一般为可复性,但当膜的损伤加重时,可经过混合型而过渡为基质型。
线粒体为对损伤极为敏感的细胞器,其肿胀可由多种损伤因子引起,其中最常见的为缺氧;此外,微生物毒素、各种毒物、射线以及渗透压改变等亦可引起。但轻度肿大有时可能为其功能升高的表现,较明显的肿胀则恒为细胞受损的表现。但只要损伤不过重、损伤因子的作用不过长,肿胀仍可恢复。
线粒体的增大有时是器官功能负荷增加引起的适应性肥大,此时线粒体的数量也常增多,例如见于器官肥大时。反之,器官萎缩时,线粒体则缩小、变少。

图1-8 心肌细胞线粒体
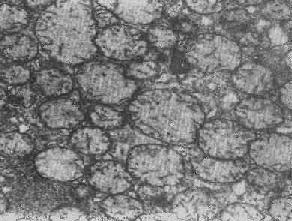
图1-9 线粒体肿
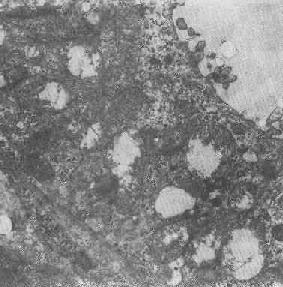
图1-10肾小管上皮细胞线粒体部分空泡变
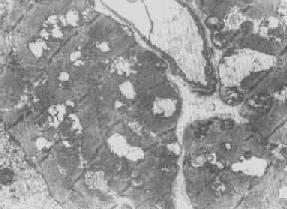
图1-11 线粒体肿胀(基质型)空泡变(心肌缺氧)
3.结构的改变线粒体嵴是能量代谢的明显指征,但嵴的增多未必均伴有呼吸链酶的增加。嵴的膜和酶平行增多反映细胞的功能负荷加重,为一种适应状态的表现;反之,如嵴的膜和酶的增多不相平行,则是胞浆适应功能障碍的表现,此时细胞功能并不升高。
在急性细胞损伤时(大多为中毒或缺氧),线粒体的嵴被破坏;慢性亚致死性细胞损伤或营养缺乏时,线粒体的蛋白合成受障,以致线粒体几乎不再能形成新的嵴。
根据细胞损伤的种类和性质,可在线粒体基质或嵴内形成病理性包含物。这些包含物有的呈晶形或副晶形(可能由蛋白构成),如在线粒体性肌病或进行性肌营养不良时所见(图1-12);有的呈无定形的电子致
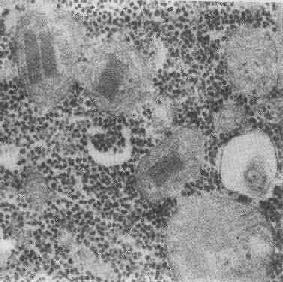
图1-12 线粒体内晶形包含体(进行性肌营养不良症)
密物,常见于细胞趋于坏死时,乃线粒体成分崩解的产物(脂质和蛋白质),被视为线粒体不可复性损伤的表现。线粒体损伤的另一种常见改变为髓鞘样层状结构的形成,这是线粒体膜损伤的结果。
衰亡或受损的线粒体,最终由细胞的自噬过程加以处理并最后被溶酶体酶所降解消化。
五、高尔基体
高尔基体(Golgi apparatus)见于一切有核细胞,来自核膜外层,由数列弯曲成蹄铁状的扁平囊组成,在横切面上表现为光面双膜,其末端膨大成烧瓶状。高尔基体面向核的一面称为形成面,由许多与粗面内质网池相连的小泡构成。另一面称为成熟面,由此断下一些较大的泡,内含分泌物。由粗面内质网合成的蛋白质输送到此,经加工装配形成分泌颗粒,分泌到细胞外,例如肝细胞合成的白蛋白和脂蛋白即按此方式形成和输出。此外,细胞本身的酶蛋白如溶酶体的水解酶类也是这样,但却不装配成分泌颗粒和排出细胞外,而是以高尔基小泡的形式(初级溶酶体,前溶酶体)输送到各种吞噬体中。高尔基体在形成含糖蛋白的分泌物中、在构成细胞膜及糖萼中,以及在形成结缔组织基质中也均起着重要的作用。
高尔基体的病变
1.高尔基体肥大高尔基体肥大见于细胞的分泌物和酶的产生旺盛时。巨噬细胞在吞噬活动旺盛时,可见形成许多吞噬体、高尔基复合物增多并从其上断下许多高尔基小泡。
2.高尔基体萎缩在各种细胞萎缩时可见高尔基体变小和部分消失。
3.高尔基体损伤时大多出现扁平囊的扩张以及扁平囊、大泡和小泡崩解。
六、溶酶体
溶酶体(lysosome)为细胞浆内由单层脂蛋白膜包绕的内含一系列酸性水解酶的小体。形态学上只有联合运用电镜和细胞化学方法才能肯定地加以确认。但是在胞浆中有一系列来源不同的小体符合这一定义,故可将溶酶体区分为以下不同的类型。
1.初级溶酶体为除水解酶类外不含其他物质并尚未参与细胞内消化过程的溶酶体,例如中性粒细胞中的嗜天青颗粒、嗜酸性细胞中的颗粒以及巨噬细胞和一些其他细胞中的高尔基小泡(图1-13)。
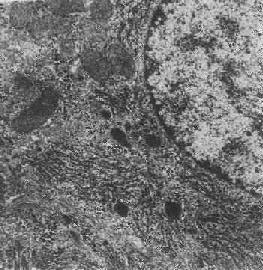
图1-13 初级溶酶体
图中央及中下方之卵圆形电子致密小体,外围单层包膜。(图中及下部片层状膜性结构为粗面内质网(正常肝细胞)
2.次级溶酶体为除溶酶体的水解酶外尚含有其他外源性或内源性物质并已参与细胞内消化过程的溶酶体(图1-14),亦即含有溶酶体酶的各种噬体,因而称为吞噬溶酶体(phagolysosome),乃由吞噬体与初级或次级溶酶体融合而成。
溶酶体是极为重要的细胞器,能与细胞的一系列生物功能和无数的物质代谢过程。因此,其功能障碍将导致细胞的病理改变,从而在许多疾病的发病机制中具有重要意义。
溶酶体的病变
1.溶酶体的病理性 贮积过程在某些病理情况下,一些内源性或外源性物质可在溶酶体内贮积,使病酶体增大和数目增多。
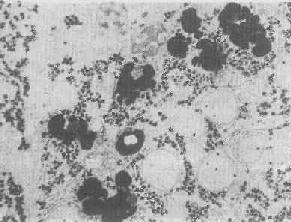
图1-14 肝细胞内次级溶酶体
贮存在溶酶体中的物质被溶酶体酶加以降解(消化)。但有时进入细胞的物质为量过多,超过了溶酶体的处理能力,于是乃在细胞内贮积,例如各种原因引起的蛋白尿时可在肾近曲小管上皮细胞中见到玻璃滴状蛋白质的贮积(所谓玻璃样小滴变性)。在电镜下可见这种玻璃样小滴乃载有蛋白质的增大的溶酶体,故实质上这往往是细胞功能增强的表现,与真正的变性有所不同。
一些在正常情况下可被消化的物质如糖原和粘多糖等,当溶酶体有先天性酶缺陷时,也能在溶酶体中堆积,如Ⅱ型糖原贮积病(Pompe)病。
2.溶酶体在细胞自溶过程中的作用 溶酶体因含有许多种水解酶,故在细胞的自溶过程中起着重要的作用。在溶酶体膜损伤及通透性升高时,水解酶逸出,引起广泛的细胞自溶。这就是活体内细胞坏死和机体死后自溶的主要过程。在此过程中,受损细胞的大分子成分被水解酶分解为小分子物质。
比细胞的广泛坏死或自溶更为重要的是溶酶体在细胞的局灶性坏死中所起的作用。此时在胞浆内形成自噬泡,在与溶酶体结合形成自噬溶酶体。如水解酶不能将其中的结构彻底消化溶解,则自噬溶酶体乃常转化为细胞内的残存小体,如某些长寿细胞中的脂褐素(图1-15)。
3.溶酶体在细胞间质损伤中的作用 当溶酶体酶释放到细胞间质中时,同样发挥其酶解破坏作用。这在诸类风湿性关节炎等炎症过程和肿瘤细胞侵入血管的过程中具有重要意义。但溶酶体酶逸出溶酶体进入细胞间隙的机制尚不十分清楚,可能由于溶酶体膜和细胞的失稳或通过出胞过程而实现的。因此,临床上可用溶酶体膜稳定剂治疗有关疾病。
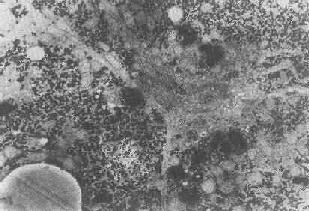
图1-15 肝细胞内脂褐素颗粒残存小体即终末溶酶体
七、过氧化
过氧化(peroxisome)为胞浆中由单层界膜包绕的另一类小体,直径为0.5~1μm,形态与细胞化学特性均不同于溶酶体。小体基质电子密度中等,中央大多含有一电子密度较大的有时呈晶状的核芯。此小体不含水解酶而含有若干种氧化酶,还有大量呈过氧化作用的触酶,被视为过氧体的标志酶。过氧体的功能至今尚不甚清楚,看来可能与糖原异生和分解有害于细胞的H2O2及脂质代谢有关。
过氧体的病变
在人体病理学方面关于过氧体的病变知之尚少。
1.过氧体增多 动物实验中,在切除甲状腺、给予皮质激素或氨基水杨酸后以及在实验性致癌过程中,可见过氧体增多。在人体的某些病理过程如某些炎症(病毒性肝炎、螺旋体感染)及慢性酒精中毒等时也可见到过氧体增多现象。
2.过氧体减少或缺如 在较罕见的脑肝肾综合症(Zellweger综合征)时,曾见到过氧体缺如的现象,但其病理意义尚不清楚。
3.微过氧体 为一组同样含有过氧化物酶和触酶的小体,但远较过氧化为小(0.15~0.25μm),不含核芯结构,与光面内质网相连。因此被认为是光面内质网的特化部分及过氧体的前身,其作用尚未查明。
八、细胞骨架
细胞骨架乃胞浆中一组由纤维状结构组成的网架,具有支撑和维持细胞形态及细胞运动的功能。迄今已知的成分有微丝、微管和中间丝3种。微丝粗约6nm,根据其生化和免疫细胞化学特性实属肌动蛋白(actin)细丝;微管为直径约20~26nm的长度不一的小管,管壁由13根纵列的原丝构成;中间丝的直径在微丝和微管之间(7~11nm)故名。
细胞骨架中的中间丝化学性质各异,在不同细胞由不同的蛋白质和多肽组成:在上皮细胞为前角蛋白或细胞角蛋白(cytokeratin),在间叶性细胞为波形蛋白(vimentine),在神经细胞为神经原丝(neurofilament),在肌细胞为桥连蛋白(desmine),在神经胶质细胞为胶质纤维酸性蛋白(gilal fibrillary acidic protein,GFAP)。 由于这些不同种类不同性质的中间丝在细胞转化为肿瘤细胞时,仍不改变其化学和抗原特异性,故可利用这种特性借助免疫细胞化学方法,对肿瘤进行分类和鉴别诊断。
关于细胞骨架在细胞损伤时的改变,所知不多,但早已知道秋水仙碱能抑制微管形成,引起有丝分裂障碍;通过对微管(可能还有微?
第三节 细胞和组织的适应性反应
细胞和组织在对各种刺激因子和环境改变进行适应时,能发生相应的功能和形态改变。
一、肥大
细胞、组织和器官体积的增大称为肥大(hypertrophy)。肥大细胞的线粒体总体积增大,细胞的合成功能升高,同时粗面内质网及游离核蛋白体增多。当酶合成增加时,光面内质网也相应增多。在功能活跃的细胞(特别是吞噬中的细胞)溶酶体也增多增大。在横纹肌功能负荷加重时,不仅线粒体、粗面内质网等细胞器及游离核蛋白体增多,肌丝也相应增多。此外,细胞核的DNA含量增加,导致核的增大和多倍体化,核形不规则。
肥大可分为代偿性肥大与内分泌性肥大二类。
1.代偿性肥大代偿性肥大通常系由相应器官的功能负荷加重引起,例如经锻炼的骨骼肌、高血压引起的心肌肥大以及一侧肾摘除后另侧肾的肥大等。
2.内分泌性肥大由内分泌作用引起的肥大,如雌激素影响下的妊娠子宫等。
二、增生
由于实质细胞数量增多而造成的组织、器官的体积增大称为增生(hyperplasia)。增生细胞的各种功能物质如细胞器和核蛋白体等并不或仅轻微增多。细胞增生是由于各种原因引起的有丝分裂活动增强的结果,通常为可复性的,当原因消除后又可复原。
增生通常有如下各种类型:
1.再生性增生 具再生能力的组织当发生严重损伤时,可通过细胞再生而修复,使之在结构上和功能上均恢复原状。例如肝细胞毒性损伤后的再生,肾小管坏死后的再生以及溶血性贫血的骨髓增生等,均属再生性增生。
2.过再生性增生 体内某些常发生慢性反复性组织损伤的部位,由于组织的反复再生修复而逐渐出现过度的增生,如慢性胃炎时的上皮腺样增生、宫颈阴道部糜烂时的上皮增生和慢性肝细胞增生等。此型增生伴有细胞的异型性并进一步转化为肿瘤细胞,例如宫颈糜烂可发展为宫颈癌、慢性肝炎可发展为肝细胞癌等。
3.内分泌障碍性增生 某些器官由于内分泌障碍可引起增生。如缺碘时可能过反馈机制障碍引起甲状腺增生,妊娠时的垂体增生,雌激素过多时的子宫内膜增生、乳腺增生等。
三、萎缩
组织和器官的体积缩小称为萎缩(atrophy),通常是由于各该组织、器官的实质细胞体积缩小造成的,有时也可因细胞数目减少引起。最常见的萎缩有肌肉、骨骼、中枢神经及生殖器官等的萎缩。
萎缩通常由于细胞的功能活动降低、血液及营养物质供应不足,以及神经和(或)内分泌刺激减少等引起。萎缩细胞的细胞器减少甚至消失,如长期不动的骨骼肌,其肌原纤维常大量消失,以致仅留下互相靠近的细胞核,貌似肌细胞核增多。肌细胞及其他实质细胞细胞器的解体大多在自噬溶酶体内进行,因而在长期活动减少和变小的肌细胞及某些其他实质细胞内,常可见到许多溶酶体性的残存小体,亦即光学显微镜下的脂褐素颗粒。
细胞和器官除体积变小外,质地常变得较坚韧,边缘变锐,色泽变深(如心和肝的褐色萎缩)。
细胞和器官发生萎缩的原因多种多样,但均含有环境条件变坏的因素,从而引起细胞和器官的体积缩小及功能下降。根据病因,可将萎缩概括地分为二大类即生理性萎缩及病理性萎缩:
1.生理性萎缩 许多结构、组织和器官当机机发育到一定阶段时乃逐渐萎缩,这种现象称为退化,例如在幼儿阶段动脉导管和脐带血管的萎缩退化、青春期后胸腺的逐步退化、妊娠期后子宫的复旧,以及授乳期后乳腺组织的复旧等。此外,在高龄时期几乎一切器官和组织均不同程度地出现萎缩,即老年性萎缩,尤以脑、心、肝、皮肤、骨骼等为明显。
2.病理性萎缩 乃在病理状态下出现的萎缩,原因不一。有的表现为全身性萎缩,有的则表现为局部性萎缩。
(1)全身性萎缩:如长期营养不良或消化道梗阻引起的饥饿性萎缩,全身消耗性疾病及恶性肿瘤患者的全身性萎缩(恶病质)等。
(2)局部性萎缩:乃在某些局部因素影响下发生的局部组织和器官的萎缩。例如动脉硬化症引起的肾萎缩、脑萎缩;长期压迫引起的压迫性萎缩(如肾盂积水时的肾实质萎缩)(图1-16);肢体、器官等长期不活动所致的废用性萎缩(如小儿麻痹症所致的下肢萎缩)以及内分泌和功能低下所至的内分泌萎缩(如Simmond病时,由于垂体功能低下引起的甲状腺、肾上腺、性腺等器官的萎缩),等等。
萎缩的后果:萎缩一般是可复性的。只要萎缩的程度不十分严重,当原因消除后,萎缩的器官、组织、细胞仍可逐渐恢复原状。但病变如继续进展,则萎缩的细胞可最后消失。
四、化生
一种已分化组织转化为另一种相似性质的分化组织的过程称为化生(metaplasia)。但这种转化过程并非表现为已分化的细胞直接转变为另一种细胞,而系由具有分裂能力的未分化细胞向另一方向分化而成,并且只能转化为性质相似的而不能转化为性质不同的细胞,例如上皮细胞不能转化为结缔组织细胞或相反。故柱状上皮可转化为鳞状上皮,一种间叶性组织只能转化为另一种间叶性组织。
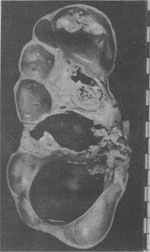
图1-16 肾压迫萎缩肾盂乳头状瘤,引起肾盂积水进而压迫肾实质引起萎缩
较常见的化生有:
1.鳞状上皮化生 常见于气管和支气管粘膜。当此处粘膜上皮时间受化学性刺激气体或慢性炎症损害而反复再生时,乃可能出现化生,即由原来的纤毛柱状上皮转化为鳞状上皮。这是一种适应性表现,通常仍为可复性的。但若持续存在,则有可能成为常见的支气管鳞状细胞癌的基础。这可能是由于纤毛上皮消失,粘膜失去其净化功能,以致致癌物质不能被及时排除的缘故。鳞状上皮化生尚可见于其他器官,如慢性胆囊炎及胆石症时胆囊粘膜上皮的鳞状上皮化生;慢性宫颈炎时的宫颈粘膜上皮的鳞状化生等。
2.肠上皮化生 这种特殊类型的化生常见于胃。此时,胃体和(或)胃窦部的粘膜腺体消失,表面上皮的增生带由胃小凹移位于粘膜基底部并改变其分化方向而分化出的小肠或大肠型粘膜上皮。这种情况常见于慢性萎缩性胃炎伴粘膜腺体消失或胃溃疡及胃糜烂后粘膜再生时。这种肠上皮化生也可成为肠型胃癌的发生基础。
3.结缔组织和支持组织化生 许多间叶性细胞常无严格固定的分化方向,故常可由一种间叶性组织分化出另一种间叶性组织。这种情况也多为适应功能改变的结果,例如,间叶组织在压力作用下可转化为透明软骨组织;有时并可发展出骨组织,例如骨骼肌反复外伤后(如骑士的缝匠肌)可在肌组织内形成骨组织,在骨化性肌炎(myositis ossificans)时也是如此。这是由于新生的结缔组织细胞转化为骨母细胞的结果。
有人认为,机体的一切真核细胞均具有相同的遗传信息,故由一种组织化生出另一种组织是可以理解的。
第四节 细胞和组织的损伤
细胞和组织损伤的表现形式和轻重程度不一,轻者当招致损伤的原因消除后仍可恢复,重者则可引起细胞和组织的死亡,兹分述如下:
一、变性和物质沉积
所谓变性(degeneration)乃指细胞或细胞间质的一系列形态改变并伴有结构和功能的变化(功能下降),表现为细胞间质内出现异常物质或正常物质而数量显着增多。但使用这一定义时必须慎重,因为有时细胞内某种物质的增多恰属生理现象而并非病理性改变。
一般而言,变性是可复性改变,当原因消除后,变性细胞的结构和功能仍可恢复。但严重的变性则往往不能恢复而发展为坏死。
变性可概括分为二大类;细胞含水量异常;细胞内物质的异常沉积。
(一)细胞水肿
人体约一半由水构成,其中约2/3存在于细胞内,约1/3存在于细胞外。在正常情况下细胞内外的水分互相交流,协调一致,保持着机体内环境的稳定。但当因缺氧、缺血、电离辐射以及冷、热、微生物毒素等的影响,而致细胞的能量供应不足、细胞膜上的钠泵受损,使细胞膜对电解质的主动运输功能发生障碍,或细胞膜直接受损时,则导致细胞内水分增多,形成细胞水肿,严重时称为细胞的水变性(hydropic degeneration)。
形态学:水肿的细胞体积增大,胞浆基质内水分含量增多,变得较为透明、淡染,胞核也常常被波及而增大、染色变淡、从而使整个细胞膨大如气球,故有气球变之称(图1-17)。电镜下,除可见胞浆基质疏松变淡外,尚可见线粒体肿胀及嵴变短、变少甚至消失;内质网则广泛解体、离断和发生空泡变。严重的水变性有时与渐进性坏死和坏死难以区分。
在电镜技术问世之前,有混浊肿胀之称,用以形容细胞肿胀和胞浆出现颗粒而变混浊,相应的器官(如心、肝、肾等实质性器官)在肉眼观上肿胀、边缘变钝、透明度降低(混浊)。电镜检查表明,胞浆内的颗粒实乃水肿时肿大的线粒体和扩张断裂的内质网,但有时也可为小泡状的蛋白质或其他物质的沉积。故为避免误解,现已放弃混浊肿胀这一名称。
结果:细胞水肿通常为细胞的轻度或中等度损伤的表现,当原因消除后仍可恢复正常。但如进一步继续发展,则可能形成脂肪变性甚或坏死。
(二)细胞内物质沉积
在病理状态下,不同的原因可以导致多种不同的物质在细胞和间质内异常沉积,为变性的主要表现。
1.脂肪沉积正常情况下,除脂肪细胞外,其他细胞内一般不见或仅见少量脂滴。如这些细胞中出现脂滴或脂滴明显增多,则称为脂肪变性(fatty degeneration)。电镜下可见脂滴形成于内质?
■[此处缺少一些内容]■
碳水化合物等结合,形成细胞的结构成分,即成为结构脂肪。因此,上述过程中的任何一个环节发生障碍便能导致肝细胞的脂肪变性;①脂蛋白合成障碍,以致不能将脂肪运输出去,造成脂肪在肝细胞内堆积。这常系由于合成脂蛋白的原料如磷脂或组成磷脂的胆碱等物质缺乏,或由于化学毒物(如酒精、四氯化碳)或其他毒素(如霉菌毒素)破坏内质网结构或抑制某些酶的活性,使脂蛋白及组成脂蛋白的磷脂、蛋白质等的合成发生障碍所致。②中性脂肪合成过多。这往往是由于饥饿或某些疾病(如消化道疾病)造成饥饿状态,或糖尿病患者对糖的利用障碍时,从脂库动员出大量脂肪,其中大部分以脂肪酸的形式进入肝,致肝合成脂肪增多,超过了肝将其氧化利用和合成脂蛋白输送出去的能力,于是导致脂肪在肝内的蓄积。③脂肪酸的氧化障碍,使细胞对脂肪的利用下降。例如白喉外毒素等能干扰脂肪酸的氧化过程,而缺氧即影响脂蛋白的合成,又影响脂肪酸的氧化。总之,肝细胞的脂肪变性乃上述某一因素或几种因素综合利用的结果。
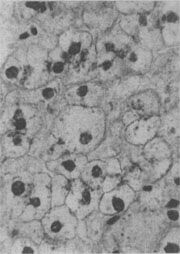
图1-17 肝细胞气球样变
病毒性肝炎时,肝细胞明显肿胀,胞浆疏松呈气球样
形态学:轻度肝脂肪变性时,肝肉眼观可无明显改变,或仅轻微黄染。如脂仿变性比较显著和广泛,则肝增大,色变黄,触之质如泥块并有油腻感。镜下,肝细胞内的脂肪空泡较小,起初多见于核的周围,以后变大,较密集散布于整个胞浆中,严重时可融合为一个大空泡,将细胞核挤向胞膜下,状似脂肪细胞(图1-18)。脂肪变性在肝小叶中的分布与其病因有一定的关系,例如肝淤血时,小叶中央区缺氧较重,故脂肪变性首先在此处发生。但长期淤血后,小叶中央区的肝细胞大多萎缩、变性或消失,于是小叶周边区肝细胞也因缺氧而发生脂肪变性。磷中毒时,肝细胞脂肪变性则主要发生于小叶周边区,这可能是由于此区肝细胞对磷中毒更为敏感的缘故。
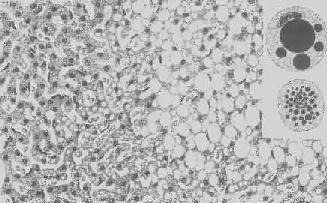
图1-18 肝细胞脂肪变性
肝细胞胞浆内出现大小不等的脂肪空泡;右上角为饿酸染色的脂肪细胞, 脂滴染成黑色
(2)心肌脂肪变性:心肌在正常情况下可含有少数脂滴,脂肪变性时脂滴明显增多。镜下,脂肪空泡较细小,呈串珠状成排排列,主要位于肌纤维Z带附近和线粒体分布区。常为贫血和中毒的结果。在严重贫血时,可见心膜下尤其是乳头肌处出现成排的黄色条纹,与正常心肌的暗红色相间排列,状若虎皮斑纹,故有“虎斑心”之称。严重感染、白喉外毒素以及其他毒物(如磷、砷、氯仿等)也能引起心肌的弥漫性脂肪变。肉眼观,心肌均匀变浊,略呈黄白色。但通常心功能并不受明显影响。显著的心肌脂肪变性如今并不常见。
(3)肾脂肪变性;在严重贫血、缺氧和中毒过程中,或肾小球毛细血管通透性升高时,肾小管特别是近曲小管的上皮细胞可吸收漏出的脂蛋白而导致脂肪变性。脂滴起初多位于细胞基底部。肉眼观,肾稍肿大,切面上可见皮质增厚,略呈浅黄色。
2.玻璃样变性又称透明变性(hyaline degeneration),为十分常见的变性,主要见于结缔组织、血管壁,有时也可见于细胞内。
1.结缔组织玻璃样变:常见于纤维瘢痕组织、纤维化的肾小球,以及动脉粥样硬化的纤维性瘢块等。此时纤维细胞明显变少,胶原纤维增粗并互相融合成为梁状、带状或片状的半透明均质,失去纤维性结构(图1-19)。质地坚韧,缺乏弹性。玻璃样变的发生机制尚不甚清楚,有人认为在纤维瘢痕老化过程中,原胶原蛋白分子的交联增多,胶原原纤维也互相融合,其间并有较多的糖蛋白积聚,形成所谓玻璃样物质;也有人认为可能由于缺氧、炎症等原因,造成局部pH升高或温度升高,致使原胶原蛋白分子变性成明胶并互相融合所致。
2.血管壁玻璃样变:这种改变常见于高血压病时的肾、脑、脾及视网膜的细动脉。此时,可能是由于细动脉的持续性痉挛,使内膜通透性增高,血浆蛋白得以渗入内膜,在内皮细胞下凝固成无结构的均匀红染物质。此外,内膜下的基底膜样物质增多。这些改变使细动脉的管壁增厚、变硬,管腔变狭,甚至闭塞(图1-20),此即细动脉硬化症(arteri-olosclerosis),可引起肾及脑的缺血。
3.细胞内玻璃样变:亦称为细胞内玻璃样小滴变性。这种情况常见于肾小球肾炎或其他疾病而伴有明显蛋白尿时。此时肾近曲小管上皮细胞胞浆内可出现许多大小不等的圆形红染小滴(图1-21),这是血浆蛋白经肾小球滤出而又被肾小管上皮细胞吞饮的结果,并在胞浆内融合成玻璃样小滴,以后可被溶酶体所消化。此外,在酒精中毒时,肝细胞核周胞浆内亦可出现不甚规则的红染玻璃样物质。电镜下,这种物质由密集的细丝构成,据认为可能是细胞骨架中含角蛋白成分改变的结果,并被称为Mallory小体。
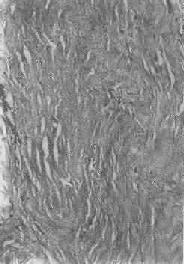
图1-19 纤维瘢痕组织的玻璃样变
胶原纤维变粗,互相融合呈均质梁状
3.纤维素样变性(纤维蛋白样变性)为间质胶原纤维及小血管壁的一种变性。病变部位的组织结构逐渐消失,变为一堆境界不甚清晰的颗粒状、小条或小块状无结构物质,呈强嗜酸性红染,状似纤维素,并且有时呈纤维素染色,故称此改变为纤维素样变性(fibrinoid degeneration),其实为组织坏死的一种表现,因而也称为纤维素样坏死(fibrinoid necrosis)。
纤维素样变性主要见于急性风湿病及结节性动脉周围炎等变态反应性疾病。至于所谓纤维素样物质的性质和形成机制,至今尚无统一意见。一般认为,在早期,结缔组织基质中有PAS阳性的粘多糖增多,以后纤维崩解为碎片,从而失去原来的组织结构而变为纤维素样物质。此外,还有免疫球蛋白增多,有时还有纤维蛋白增多。这种改变可能是抗原抗体反应时形成的生物活性物质使间质受损、胶原纤维崩解所致。同时,附近小血管也可受损,引起通透性升高、血浆渗出,并在组织凝血系统的酶的催化作用下,使血浆纤维蛋白原转化为纤维蛋白。
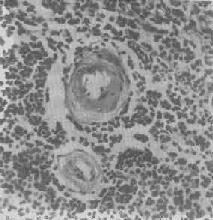
图1-20 脾中央动脉玻璃样变
中央动脉管壁明显增厚,呈玻璃样均质状,管腔变窄
4.粘液性变性组织间质内出现类粘液的积聚称为粘液样变性(mucoid degeneration)。镜下可见病变处的间质变疏松,充以染成淡蓝色的胶状液体,其中有一些多角形、星芒状细胞散在,并以突起互相联缀。
结缔组织粘液样变性常见于间叶性肿瘤、急性风湿病时的心血管壁、动脉粥样硬化的血管壁。甲状腺功能低下时,全身皮肤的真皮及皮下组织的基质中有较多类粘液(mucoid)及水分潴留,形成粘液水肿(myxedema),其机制可能是甲状腺功能低下时,能促进透明质酸酶活性的甲状腺素分泌减少,致透明质酸(类粘液的主要成分之一)降解减弱而大量潴积于组织内。
粘液样变性当病因消除后可以逐渐消退,但如长期存在,则可引起纤维组织增生,从而导致组织硬化。
5.淀粉样变性组织内有淀粉样物质沉积称为淀粉样变性(amyloid degeneration,amyloidosis)。淀粉样物质为一种结合粘多糖的蛋白质,遇碘时被染成赤褐色,再加以硫酸则呈蓝色,与淀粉遇碘时的反应相似,故称之为淀粉样物质。此物质常浸润于细胞间或沉积于小血管的基底膜下,或沿网状纤维支架分布(图1-22)。淀粉样物质在HE染色切片中为淡红色均质状,电镜下则为纤细的丝状。
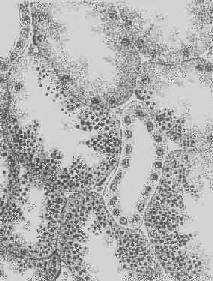
图1-21 肾近曲小管上皮细胞玻璃样小滴变
上皮细胞胞浆内出现大小不等的均质圆形小滴

图1-22 肝淀粉样变
淀粉样物质呈均质状,沉着于窦内皮下,肝细胞索受压萎缩
淀粉样变可为全身性和局部性二种。前者在我国极为罕见,多发生在长期慢性化脓、骨髓瘤及结核病等情况下。局部性淀粉样变则较常见,并好发于睑结膜及上呼吸道等处的慢性炎症而伴有大量浆细胞浸润时,发生机制不清。根据淀粉样物质中常有丙种球蛋白和血清中球蛋白增多的现象,有人认为这是由于抗原抗体反应在血中形成的蛋白复合物,也有人认为是浆细胞产生的免疫球蛋白与纤维母细胞、内皮细胞所产生的含硫粘多糖相结合而形成的复合物。
6.病理性色素沉积组织中可有各种色素沉积,其中有的来源于机体自身,称为内源性色素,如含铁血黄素、胆色素、脂褐素、黑色素等;有的则来自体外,为外源性色素,如炭末及纹身所用的色素。常见的病理性色素沉积有以下几种:
(1)含铁血黄素:含铁血黄素(hemosiderin)是由铁蛋白(ferritin)微粒集结而成的色素颗粒,呈金黄色或棕黄色而具有折光性。颗粒大小不一,是巨噬细胞吞食红细胞后在胞浆内形成的一种色素,为血红蛋白被巨噬细胞溶酶体分解而转化所成。由于铁蛋白分子中含有高铁(Fe3+),故遇铁氰化钾及盐酸后出现蓝色反应,称为普鲁士蓝或柏林蓝色反应。细胞破裂后,此色素亦可散布于组织间质中。左心衰竭时,在发生淤血的肺内可有红细胞漏出肺泡中,被巨噬细胞吞噬后形成含铁血黄素。这种细胞可出现于患者痰内,即所谓心衰细胞。当溶血性贫血时有大量红细胞被破坏,可出现全身性含铁血黄素沉积,主要见于肝、脾、淋巴结、骨髓等器官。
(2)胆红素(bilirubin):也是在吞噬细胞内形成的一种血红蛋白衍生物。在生理情况下,衰老的红细胞在单核吞噬细胞中被破坏,其血红蛋白被分解为珠蛋白、铁及胆绿素,后者还原后即成为胆红素,进入血液。血中胆红素过多时则将组织染成黄色,称为黄疸。胆红素一般呈溶解状态,但也可为黄褐色折光性小颗粒或团块。在胆道阻塞及某些肝疾患时,肝细胞、毛细胆管及小胆管内可见许多胆红素。黄疸明显时,胆红素颗粒亦可见于Kupffer细胞、肾小管上皮细胞内,并可在肾小管腔内形成胆汁管型。但人体因有血脑屏障的保护,胆红素通常不能进入脑和脊髓,而在新生儿则由于血脑屏障尚不完善,故在高胆红素血症(hyperbilirubinemia)时,大量胆红素可进入脑细胞内,使其氧化磷酸化过程受障,能量产生受抑,细胞乃发生变性,引起神经症状。肉眼观可见多处神经核(豆状核、下丘脑、海马回等)明显黄染,故称之为核黄疸。
(3)脂褐素(lipofuscin):为一种黄褐色细颗粒状色素,其成分约50%为脂质,其余为蛋白质及其他物质。脂褐素颗粒为细胞内自噬溶酶体(autophagolysosome)中的细胞器碎片发生某种理化改变后,不能被溶酶体酶所消化而形成的一种不溶性残存小体。正常人的附睾上皮细胞、睾丸间质细胞以及某些神经细胞的胞浆中可含有少量脂褐素。老年人及一些慢性消耗性疾病患者的肝细胞、肾上腺皮质网状带细胞的胞浆以及心肌细胞核两侧的胞浆中,均可出现脂褐素,故此色素又有消耗性色素之称。脂褐素颗粒在电镜下呈典型的残存小体(residual bodies)结构。
(4)黑色素(melanin):为大不、形状不一的棕褐色或深褐色颗粒色素。正常人皮肤、毛发、虹膜及脉胳膜等处均有黑色素存在。皮肤黑色素由黑色素细胞(melanocyte)产生:黑色素细胞中的酪氨酸在酪氨酶的作用下,氧化为二羟苯丙氨酸(dihydroxyphenylalanine,DOPA,多巴)。多巴被进一步氧化为吲哚醌,失去CO2后转变为二羟吲哚,后者聚后成一种不溶性的聚合物,即黑色素,再与蛋白质结合为黑色素蛋白。黑色素细胞内因含有酪氨酸酶,故当加上多巴时,则出现与黑色素相似的物质,谓之多巴反应阳性;相反,表皮下的噬黑色素细胞(melanophore),即吞噬了黑色素的组织细胞,因不含酪氨酸酶,故多巴反应阴性。用此方法可以鉴别黑色素细胞和噬黑色素细胞。人的垂体所分泌的ACTH能刺激黑色素细胞,促进其黑色素形成。当肾上腺功能低下时(例如Addison病时),全身皮肤黑色素增多,这是由于肾上腺皮质激素分泌减少,对垂体的反馈抑制作用减弱,致ACTH分泌增多,促进黑色素细胞产生过多的黑色素所致。局限性黑色素增多则见于黑色素痣及黑色素瘤等。
7.病理性钙化正常机体内只有骨和牙含有固态的钙盐,如在骨和牙之外的其他部位组织内有固态的钙盐沉积,则称为病理性钙化。沉积的钙盐主要是磷酸钙,其次为碳酸钙。
在HE染色时,钙盐呈蓝色颗粒状,开始时颗粒微细,以后聚集成较大颗粒或片块,量多时肉眼可见为白色石灰样质块,难以完全吸收而成为机体内长期存在的异物,并刺激周围结缔组织增生而将其包裹。
病理性钙化主要有营养不良性钙化和转移性钙化两种。前者颇常见,乃变性坏死组织或异物的钙盐沉积,如结核坏死灶、脂肪坏死灶、动脉粥样硬化斑块内的变性坏死区,坏死的寄生虫虫体、虫卵以及其他异物等。此时,因无全身性钙磷代谢障碍,故血钙不升高。
营养不良性钙化的机制尚未阐明,可能与局部硷性磷酸酶升高有关:碱性磷酸酶能水解有机磷酸酶,使局部磷酸增多,超过3Ca2+×2PO43-的常值,于是形成磷酸钙沉淀。至于磷酸酶的来源,一部分显然是来自坏死细胞中的溶酶体,另一部分则可能系吸收自周围组织液中的磷酸酶。此外,钙化与局部组织的pH变动有关。变性坏死组织的酸性环境可使局部钙盐溶解,钙离子浓度升高,尔后由于组织液的缓冲作用,局部组织碱性化,故钙盐乃析出沉积。
转移性钙化较少见,是全身性钙、磷代谢障碍致血钙和(或)血磷升高,使钙盐在未受损的组织上沉积所致。如甲状旁腺功能亢进及骨肿瘤造成骨质严重破坏时,大量骨钙进入血液,使血钙升高,以致钙在肾小管、肺泡和胃粘膜等处沉积,形成转移性钙化。此外,接受超剂量的维生素D时,因促进钙从肠吸收,也可引起转移性钙化。
钙化对机体的影响视具体情况而异。转移性钙化可使钙化的组织、细胞丧失;血管壁钙化使血管失去弹性变脆,容易破裂出血。但结核病灶的钙化则有可能使其中的结核菌逐渐失去活力,减少复发的危险。然而结核菌在钙化灶中往往可以继续存活很长时间,一旦机体抵抗力下降,则仍有可能引起复发。
二、坏死
生活机体的局部组织、细胞死亡后出现的形态学改变称为坏死(necrosis)。坏死组织、细胞的代谢停止,功能丧失,出现一系列特征性的形态学改变。坏死的原因多种多样,举凡一切损伤因子,只要其作用达到一定的强度或持续一定的时间,从而使受损组织、细胞的代谢完全停止时,即引起组织、细胞的死亡(坏死)。在多数情况下,坏死是由组织、细胞的变性逐渐发展而来的,即渐进性坏死(necrobiosis)。在此期间,只要坏死尚未发生而病因被消除,则组织、细胞的损伤仍可能恢复(可复期)。但一旦组织、细胞的损伤严重,代谢紊乱,出现一系列坏死的形态学改变时,则损伤不再能恢复(不可复期)。
在个别情况下,由于致病因子极为强烈,坏死可迅速发生,有时甚至无明显的形态学改变。例如将生活中的组织、细胞立即投入甲醛溶液中固定时,细胞迅即死亡,但形态上则保持完好。故单纯从形态上有时难以判断细胞是否死亡。
由于损伤因子的性质不同,引起细胞死亡的途径也各异:X线引起DNA螺旋的断裂,导致细胞核信息中心的损害;CCl4通过过氧化物阻断核蛋白合成的翻译过程;缺血阻断线粒体的呼吸链;维生素A中毒使溶酶体酶外逸。因而,根据不同的损伤类型,某种细胞器的病变乃居于主要地位。
(一)坏死的病变
细胞坏死过程中的可复性改变与不可复性改变之间并无截然的界限,只有在损伤的后期,当出现明显的形态学改变时,才能在电子显微镜下判断细胞业已死亡。而在光学显微镜下,通常要在细胞死亡后若干小时之后,当自溶性改变相当明显时才能加以辨别。
1.细胞核的改变 细胞核的改变是细胞坏死的主要形态学标志,表现为:①核浓缩(pyknosis),即由于核脱水使染色质浓缩,染色变深,核的体积缩小;②核碎裂(karyorrhexis),核染色质崩解为小碎片,核膜破裂,染色质碎片分散在胞浆中;③核溶解(karyolysis),在脱氧核糖核酸酶的作用下,染色质的DNA分解,核乃失去对碱性染料的亲和力,因而染色质变淡,甚至只能见到核的轮廓。往后染色质中残余的蛋白质被溶蛋白酶所溶解,核便完全消失(图1-23)。这一状态约经10小时才能达到正常细胞核浓缩核碎裂核溶解消失
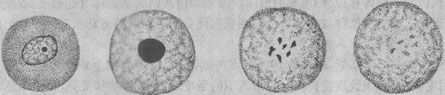
图1-23 细胞坏死时核的变化模式图
坏死细胞核的上述变化过程可因损伤因子作用的强弱和发展过程的快慢而有所不同。损伤因子的作用较弱、病变经过缓慢时(如缺血栓梗死),上述核的改变可顺序发生,即先出现核浓缩,然后碎裂,最后核溶解;但如损伤因子作用强烈,经过急剧(如中毒),则往往先发生染色质边集,继即进入核碎裂,甚或可从正常核迅即发生核溶解。
2.细胞浆的改变 坏死细胞的胞浆红染(即嗜酸性),这是由于胞浆嗜硷性物质核蛋白体减少或丧失,使胞浆与硷性染料苏木素的结合减少而与酸性染料伊红的结合力增高的缘故。同时由于胞浆结构崩解,致胞浆呈颗粒状。有时由于实质细胞坏死后,整个细胞迅速溶解、吸收而消失(溶解性坏死)。有时单个实质细胞(如肝细胞)坏死后,胞浆内水分逐渐丧失,核浓缩而后消失,胞体固缩,胞浆呈强酸性染色(红染),形成所谓嗜酸性小体,称为嗜酸性坏死或固缩坏死。
3.间质的改变实质细胞坏死后一段时间内,间质常无改变。以后在各种溶解酶的作用下,基质崩解,胶原纤维肿胀并崩解断裂或液化。于是坏死的细胞和崩解的间质融合成一片模糊的颗粒状、无结构的红染物质。
上述坏死的形态学改变通常要在组织、细胞死亡后相当时间(数小时至10小时以上)才出现。在坏死的早期阶段,不仅肉眼观难以鉴别,甚至在电子显微镜下也不能确定该组织、细胞是否死亡。临床上将这种已失去生活能力的组织称为失活组织。这种组织已不能复活,但却是细菌生长繁殖的良好基地。为防止感染,促进愈合,在治疗中常需将其清除。
(二)坏死的类型
形态学上可将坏死分为以下类型:
1.局灶性胞浆坏死 并非任何致死性的细胞损伤均必然导致整个细胞的坏死。有时坏死仅局限于细胞的某一部位,称为局灶性胞浆坏死。这种坏死区由界膜加以包裹,从而与周围健康胞浆相隔离。这样,各种水解酶就可经内质网系统进入其中,将坏死结构加以溶解消化,而不致损伤细胞的其余部分。
2.凝固性坏死 坏死组织由于失水变干、蛋白质凝固而变成灰白或黄白色比较坚实的凝固体,故称凝固性坏死(coagulation necrosis)。特点是坏死组织的水分减少,而结构轮廓则依然较长时间地保存。
凝固性坏死的发生机制仍不甚清楚。Weigert认为是胞浆凝固的结果,因为溶酶体酶在此不起重要作用,这是由于或则组织所含溶酶体较少,或则溶酶体酶在某种程序上也受到了损伤的缘故。但在细胞僵死以前,在生体显微镜下可见到胞浆内的运动过程加强,表现为线粒体的一阵阵移位、细胞膜的波浪状活动和内质网的伪足样突起形成,以及细胞核的旋转运动。细胞的这种临终期运动与细胞有丝分裂开始时的运动相同。因此可以说细胞的死亡与新生的过程是相似的。
凝固性坏死的形态学:凝固性坏死灶在开始阶段,由于周围组织液的进入而明显肿胀,透明度降低,组织纹理变模糊。尔后组织的坚度逐渐增加,状如煮熟,呈土黄色。这些改变最早要在细胞死亡开始后6~8小时以后才能见到。坏死灶的周围形成一暗红色缘(出血边带)与健康组织分界。镜下,在较早期可见坏死组织的细胞结构消失,但组织结构的轮廓依然保存。例如肾的贫血性梗死初期,虽然细胞已呈坏死改变,但肾小球、肾小管以及血管等的轮廓仍可辨认。脾的贫血性梗死也是如此。
凝固性坏死的特殊类型:
(1)干酪样坏死(caseous necrosis, caseation):主要见于结核杆菌引起的坏死,如结核病灶的坏死。这时坏死组织彻底崩解,镜下不见组织轮廓,只见一些无定形的颗粒状物质(图1-24),同时由于坏死组织含有较多脂质(来自崩解的粒细胞和结核杆菌)故略带黄色,加之脂质又阻抑了溶酶体酶的溶蛋白作用,故结果形成了状如干酪的物质,因而得名。

图1-24 干酪性坏死(镜下)
结核病灶中心的干酪样坏死,呈一片模糊细颗粒状无结构物质
(2)坏疽(gangrene):坏疽为组织坏死后又发生了继发性改变的结果。当大块组织坏死后,由于发生了不同程度的腐败菌感染和其他因素的影响而呈现黑色、污绿色等特殊形态改变,即成为坏疽。坏死组织经腐败菌分散,产生硫化氢,后者与血红蛋白中分解出来的铁相结合,乃形成黑色的硫化铁,使坏死组织呈黑色。坏疽可分为此下3种类型:
1)干性坏疽:干性坏疽是凝固性坏死加上坏死组织的水分蒸发变干的结果,大多见于四肢末端,例如动脉粥样硬化、血栓闭塞性脉管炎和冻伤等疾患时。此时动脉受阻而静脉仍通畅,故坏死组织的水分少,再加上在空气中蒸发,故病变部位干固皱缩,呈黑褐色,与周围健康组织之间有明显的分界线(图1-25)。同时,由于坏死组织比较干燥,故既可防止细菌的入侵,也可阻抑坏死组织的自溶分解。因而干性坏疽的腐败菌感染一般较轻。
2)湿性坏疽:湿性坏疽多发生于内脏(子宫、肺等),也可见于四肢(当其动脉闭塞而静脉回流又受阻,伴有淤血水肿时)。此时由于坏死组织含水分较多,适合腐败菌生长繁殖,故腐败菌感染严重,局部明显肿胀,呈深蓝、暗绿或污黑色。腐败菌分解蛋白质,产生吲哚(indole)、粪臭素(scatole)等,造成恶臭。由于病变发展较快,炎症比较弥漫,故坏死组织与健康组织的分界线不明显。同时组织坏死腐败所产生的毒性产物及细菌毒素被吸收后,可引起严重的全身中毒症状。常见的湿性坏疽有坏疽性阑尾炎、肠坏疽、肺坏疽及产后坏疽性子宫内膜炎等。
3)气性坏疽:为湿性坏死的一种特殊类型,主要见于严重的深达肌肉的开放性创伤合并产气荚膜杆菌、恶生水肿杆菌及腐败弧菌等厌气菌感染时,细菌分解坏死组织时产生大量气体,使坏死组织内含气泡呈蜂窝状,按之有捻发音。气性坏疽发展迅速,毒素吸收多,后果严重,需紧急处理。
3.液化性坏死 在液化性坏死(liquefaction necrosis)时,坏死组织起初肿胀,随即发生酶性溶解,形成软化灶。此时,坏死组织的水解占主导地位。与凝固性坏死相反,液化性坏死主要发生在含可凝固的蛋白质少和脂质多(如脑)或产生蛋白酶多(如胰腺)的组织。凝固性坏死的组织发生细菌感染时,则白细胞的水解酶也能引起组织溶解液化(如感染的肺梗死)。
液化性坏死时,坏死组织分解液化而呈液化,并可形成坏死腔。
脂肪坏死:脂肪坏死为液化性坏死的一种特殊类型,主要有酶解性脂肪坏死和外伤性脂肪坏死两种。前者常见于急性胰腺炎时,此时胰腺组织受损,胰酶外逸并被激活,从而引起胰腺自身消化和胰周围及腹腔器官的脂肪组织被胰脂酶所分解,其中的脂肪酸与组织中的钙结合形成钙皂,表现为不透明的灰白色的斑点或斑块。镜下,坏死的脂肪细胞仅留下模糊混浊的轮廓。外伤性脂肪坏死则大多见于乳房,此时由于脂肪组织受伤而致命伤脂肪细胞破裂,脂肪外逸,并常在乳房内形成肿块,镜下可见其中含有大量吞噬脂滴的巨噬细胞(泡沫细胞)和多核异物巨细胞。
4.固缩坏死 固缩坏大多为细胞的生理性死亡,乃细胞衰老过程中各个细胞功能逐渐息灭的结果。这种坏死通常仅表现为单个细胞坏死或小灶状坏死,而从不见整个实质区细胞同时坏死,故可与树叶的枯萎凋谢过程(apoptosis,凋落)相比拟。固缩坏死(凋落)的发生机制与上述凝固性或液化性坏死均不同,而是由细胞的遗传素质而决定的。根据遗传素质,各种细胞损伤性刺激可改变遗传信息的转录和(或)翻译,形成死亡蛋白,后者能激活核酸内切酶,引起核染色质的凝集,标志着固缩性坏死的开始,不可复性的胞浆损害继之发生。
固缩坏死即凋落见于许多生理和病理过程中,为各种更替性组织中细胞衰亡更新的表现,也可见于照射及应用细胞抑制剂之后和数目性萎缩时。肿瘤细胞也能自发地发生固缩坏死。
固缩坏死的最初形态表现为染色质沿皱缩的核膜下凝聚,细胞连接松解,微绒毛及细胞突起消失;细胞表面有一些泡状胞浆膨隆,可脱落形成凋落小体(图1-26),后者可见于腺腔内或被周围健康细胞所吞噬(例如肝细胞内的Councilman小体);继而内质网池扩大、断裂,线粒体结构虽仍完好,但基质呈絮状致密化。
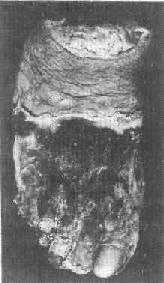
图1-25 足干性坏疽
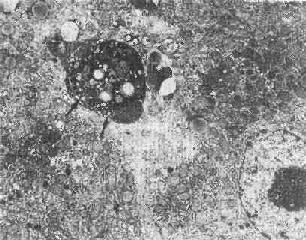
图1-26 肝细胞凋落小体(左上)
图中左上方电子致密团块(↑),其中可见脂滴及变性崩解细胞器。
其右侧为一髓鞘样结构
(三)坏死的结局
1.溶解吸收 这是机体处理坏死组织的基本方式。来自坏死组织本身和中性粒细胞的溶蛋白酶将坏死物质进一步分解、液化,然后由淋巴管或血管加以吸收,不能吸收的碎片则由巨噬细胞加以吞噬消化。留下的组织缺损则由细胞再生或肉芽组织形成予以修复或形成含有淡黄色液体的囊腔(如脑软化灶)。
2.分离排出 较大坏死灶不易完全吸收,其周围发生炎性反应,其中的白细胞释放溶蛋白酶,加速坏死边缘坏死组织的溶解吸收,使坏死灶与健康组织分离。坏死灶如位于皮肤的或粘膜,则坏死组织脱落后形成溃疡;肾、肺等内脏器官坏死组织液化后可经相应管道(输尿管、气管)排出,留下空腔,称为空洞。溃疡和空洞以后仍可修复。
3.机化(organization) 坏死组织如不能完全溶解吸收或分离排出,则由周围组织新生毛细血管和纤维母细胞等组成肉芽组织,长入坏死,逐渐加以溶解、吸收和取代,最后成为瘢痕组织。这种由新生肉芽组织取代坏死组织(或其他异物如血栓等)的过程称为机化。
4.包裹、钙化 坏死灶如较大,或坏死物质难以溶解吸收,或不能完全机化,则常由周围新生结缔组织加以包裹(encapsulation),其中的坏死物质有时可发生钙化(calcification),如结核病灶的干酪样坏死即常发生这种改变。
第五节 细胞损伤的原因和机制
引起细胞和组织损伤的原因多种多样,其作用的强弱和持续的时间决定着损伤的程度,有的引起较轻的可复性损伤,有的则引起严重的不可复性损伤,导致细胞、组织的死亡。损伤的原因可归纳为以下各类。
1.缺氧氧是细胞维持生命活动和功能的少可缺少的要素。缺氧破坏细胞的有氧呼吸,损害线粒体的氧化磷酸过程,使ATP的产生减少甚至停止,从而引起一系列的改变。缺氧可为全身性亦可为局部性,前者乃因空气稀薄(如高山缺氧)或呼吸功能障碍(如呼吸道和肺疾患),或某些化学毒物损害了血红蛋白的载氧能力(如CO中毒),或灭活呼吸链的酶系(如氰化物)所致。局部缺氧的原因则往往是缺血,常由局部循环障碍引起。
2.物理因子包括机械性、高温、低温、电流、射线等刺激因子。机械性损伤能使细胞、组织破裂;高温可使细胞内蛋白质(包括酶)变性,低温可使血管收缩、受损、引起组织缺血、细胞损害;电流通过组织时引起高温,同时也可直接刺激组织、特别是神经组织,引起功能障碍;电离射线能直接或间接造成生物大分子损伤,引起细胞损伤和功能障碍。
3.化学因子许多物质能与细胞或组织发生化学反应,从而引起细胞的功能障碍或破坏,这些物质称为毒物。其毒性作用的前提条件是毒物的可吸收性(经皮、经口或经呼吸道),其损害作用则决定于其浓度和作用持续时间。毒物的作用点或为其接触部位(如皮肤),或为其富集部位(如肺的油脂肺炎),或为其代谢部位(如肝),或为其排泄部位(如肾)。毒物进入机体的方式是,或借助于载体分子经主动运输过程进入细胞,或被动地被机体吸收。这一过程与毒物的亲水性高低呈正相关关系,而与其分子大小则呈负相关关系。进入机体后,亲水性毒物主要通过与细胞的受体相结合而损害细胞,而亲脂性毒物则主要富集于脂肪组织。
毒物的作用机制多种多样,其中最常见的是通过影响酶系统发挥其毒性作用,主要是以各种方式抑制酶的活性。此外,有些毒物则能改变血红蛋白,阻抑其运输氧的能力。还有许多毒物以及药物则抑制神经体液性刺激传导过程,或破坏遗传物质,或破坏蛋白合成,或影响免疫机构,引起过敏反应。还有许多毒物则能在其接触部位(皮肤、粘膜、肺)直接造成组织损伤。有些毒物则在机体内经过代谢才成为细胞毒,例如CCl4在肝细胞光面内质网的酶作用后才转化为具有强毒力的自由基CCl3·及Cl·,能破坏细胞的膜性结构。
4.生物因子可引起细胞损伤的生物因子有多种细菌(如白喉杆菌外毒素能抑制细胞的氧化过程和蛋白合成,链球菌溶血素能破坏细胞膜,结核杆菌通过引起变态反应造成组织损伤等),病毒(寄生在细胞内,干扰细胞的代谢过程,或产生毒性蛋白质、或通过变态反应,引起细胞和组织损伤),真菌(如放线菌、白色念珠菌、黄曲霉菌等均可以其毒素损伤组织),原虫(如疟原虫,溶组织阿米巴等),寄生虫(如日本血吸虫及其虫卵、丝虫等均可以其毒性代谢产物或分泌物引起组织损伤或通过引起反态反应造成组织损伤)。
5.免疫反应免疫反应是机体的防御功能,本身具有保护机体免患疾病的积极意义和作用。但在一定条件下,反应的结果又往往造成机体和组织的损伤,其中包括针对异体蛋白质及其他抗原而发生的变态反应如风湿热及弥漫性肾小球肾炎,以及针对自身组织发生的自身免疫反应如红斑狼疮、类风湿性关节炎等,均能造成组织损伤。
综上观之,可引起细胞、组织损伤的原因虽然十分广泛,但其基本作用机制和基本原理无非是影响与细胞基本生命活动和功能有关的细胞内的功能中心及有关细胞器:①线粒体的氧化系统,②决定合成过程的中心如内质网、核蛋白体、mRNA、核仁,③细胞的膜系统及其细胞器,④细胞核(DNA)及⑤与某些细胞的特异性功能有关的成分如肌原纤维等。
第二章 损伤的修复
损伤造成机体部分细胞作组织丧失后,机体对所形成缺损进行修补恢复的过程,称为修复(repair),修复后可完全或部分恢复原组织的结构和功能。修复过程起始于炎症,炎症渗出处理坏死的细胞、组织碎片,然后由损伤局部周围的健康细胞分裂增生来完成修复过程。修复可分为两种不同的过程及结局:①由损伤部周围的同种细胞来修复,称为再生(regeneration),如果完全恢复了原组织的结构及功能,则称为完全再生;②由纤维结缔组织来修复,称为纤维性修复。常见于再生能力弱或缺乏再生能力的组织,当其发生缺损时,不能通过原来组织再生修复,而是由肉芽组织填补,以后形成瘢痕,故也称瘢痕修复,过去常称为不完全再生。在多数情况下,由于有多种组织发生损伤,故上述两种修复过程常同时存在。本章以创伤愈合为例作进一步说明。
第一节 再生
再生可分为生理性再生及病理性再生。生理性再生是指在生理过程中,有些细胞、组织不断老化、消耗,由新生的同种细胞不断补充,始终保持着原有的结构和功能,维持着机体的完整与稳定。例如,表皮的表层角化细胞经常脱落,而表皮的基底细胞不断地增生、分化,予以补充;消化道粘膜上皮约1~2天就更新一次;子宫内膜周期性脱落,又由基底部细胞增生加以恢复;红细胞平均寿命为120天,白细胞的寿命长短不一,短的如中性粒细胞,只存活1~3天,因此不断地从淋巴造血器官输出大量新生的细胞进行补充。本章乃指病理状态下细胞、组织缺损后发生的再生,称为病理性再生。
一、组织的再生能力
各种组织有不同的再生能力,这是在动物长期进化过程中形成的。一般说来,低等动物组织的再生能力比高等动物强,分化低的组织比分化高的组织再生能力强,平常容易遭受损伤的组织以及在生理条件下经常更新的组织,有较强的再生能力。反之,则再生能力较弱或缺乏。按再生能力的强弱,可将人体组织细胞分为三类。
1.不稳定细胞(labile cells) 这类细胞总在不断地增殖,以代替衰亡或破坏的细胞,如表皮细胞、呼吸道和消化道粘膜被覆细胞、男性及女性生殖器官管腔的被覆细胞、淋巴及造血细胞、间皮细胞等。这些细胞的再生能力相当强。
2.稳定细胞(stable cells) 在生理情况下,这类细胞增殖现象不明显,似乎在细胞增殖周期中处于静止期(G0),但受到组织损伤的刺激时,则进入DNA合成前期(G1),表现出较强的再生能力。这类细胞包括各种腺体或腺样器官的实质细胞,如肝、胰、涎腺、内分泌腺、汗腺、皮脂腺和肾小管的上皮细胞等;还包括原始的间叶细胞及其分化出来的各种细胞。它们不仅有强的再生能力,而且原始间叶细胞还有很强的分化能力,可向许多特异的间叶细胞分化。例如骨折愈合时,间叶细胞增生,并向软骨母细胞及骨母细胞分化;平滑肌细胞也属于稳定细胞,但一般情况下其再生能力弱。
3.永久性细胞(permanent cells) 属于这类的细胞有神经细胞、骨骼肌细胞及心肌细胞。不论中枢神经细胞及周围神经的神经节细胞,在出生后都不能分裂增生,一旦遭受破坏则成为永久性缺失。但这不包括神经纤维,在神经细胞存活的前提下,受损的神经纤维有着活跃的再生能力。心肌和横纹肌细胞虽然有微弱的再生能力,但对于损伤后的修复几乎没有意义,基本上通过瘢痕修复。
二、各种组织的再生过程
(一)上皮组织的再生
1.被覆上皮再生 鳞状上皮缺损时,由创缘或底部的基底层细胞分裂增生,向缺损中心迁移,先形成单层上皮,以后增生分化为鳞状上皮。粘膜如胃肠粘膜上皮缺损后,同样也由邻近的基底部细胞分裂增生来修补,新生的上皮细胞起初为立方形,以后增高变为柱状细胞。
2.腺上皮再生腺上皮虽有较强的再生力,但再生的情况依损伤的状态而异:如果仅有腺上皮的缺损而腺体的基底膜未被破坏,可由残存细胞分裂补充,完全恢复原来腺体结构。如腺体构造(包括基底膜)被完全破坏,则难以再生。构造比较简单的腺体如子宫腺、肠腺等可从残留部细胞再生。肝细胞有活跃的再生力,肝再生可分为三种情况:①肝大部分切除后,剩余的肝细胞分裂增生十分活跃,短期内就能使肝恢复原来的大小。例如大白鼠肝切除90%后,只需2周就可恢复原肝的重量,不过以后要经过较长时间的结构改建,形成新的肝小叶,才能恢复原结构。②肝细胞坏死时,不认范围大小,只要肝小叶网状支架完整,从肝小叶周边区再生的肝细胞可沿支架延伸,恢复正常结构。③肝细胞坏死较广泛,肝小叶网状支架塌陷,网状纤维转化为胶原纤维(网状纤维胶原化),或者由于肝细胞反复坏死及炎症刺激,纤维组织大量增生,形成肝小叶内间隔,此时再生肝细胞难以恢复原来小叶结构,成为结构紊乱的肝细胞团,例如肝硬变时的再生结节。
(二)纤维组织的再生
在损伤的刺激下,受损处的纤维母细胞进行分裂、增生。纤维母细胞可由静止状态的纤维细胞转变而来,或由未分化的间叶细胞分化而来。幼稚的纤维母细胞胞体大,两端常有突起,突起亦可呈星状,胞浆略显嗜碱性。电镜下,可见胞浆内有丰富的粗面内质网及核蛋白体,说明其合成蛋白的功能很活跃;胞核体积大,染色淡,有1~2个核仁。当纤维母细胞停止分裂后,开始合成并分泌前胶原蛋白,在细胞周围形成胶原纤维,细胞逐渐成熟,变成长梭形,胞浆越来越少,核越来越深染,成为纤维细胞(图2-1)。
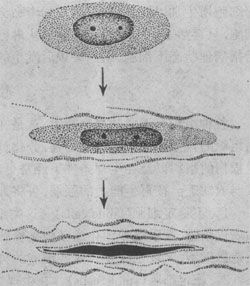
图2-1 纤维母细胞产生胶原纤维并转化为纤维细胞模式图
(三)软骨组织和骨组织的再生
软骨组织再生起始于软骨膜的增生,这些增生的幼稚细胞形似纤维母细胞,以后逐渐变为软骨母细胞,并形成软骨基质,细胞被埋在软骨陷窝内而变为静止的软骨细胞。软骨再生力弱,软骨组织缺损较大时由纤维组织参与修补。
骨组织再生力强,骨折后可完全修复。
(四)血管的再生
1.血管的再生 毛细血管多以生芽方式再生。首先在蛋白分解酶作用下基底膜分解,该处内皮细胞分裂增生形成突起的幼芽,随着内皮细胞向前移动及后续细胞的增生而形成一条细胞索,数小时后便可出现管腔,形成新生的毛细血管,进而彼此吻合构成毛细血管网(图2-2)。增生的内皮细胞分化成熟时还分泌IV型胶原、层粘连蛋白和纤维粘连蛋白,形成基底膜的基板。纤维母细胞分泌Ⅲ型胶原及基质,组成基底膜的网板,本身则成为周细胞(即血管外膜细胞)。至此毛细血管的结构逐告完成。新生的毛细血管基底膜不完整,内皮细胞间空隙较多较大,故通透性较高。为适应功能的需要,这些毛细血管还会不断改建:有的管壁增厚发展为小动脉、小静脉,其平滑肌等成分可能由血管外未分化间叶细胞分化而来。
2.大血管的修复 大血管离断后需的手术吻合,吻合处两侧内皮细胞分裂增生,互相连接,恢复原来内膜结构。但离断的肌层不易完全再生,而由结缔组织增生连接,形成瘢痕修复。
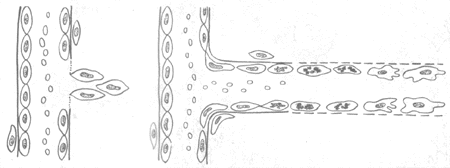
图2-2 毛细血管再生模式图
1.基底膜分解,内皮细胞肥大、增生,形成幼芽
2.内皮细胞向前移动,其后的内皮细胞分裂增生,靠近血管处的内皮细胞先分化成熟,并有新的基底膜形成
(五) 肌组织的再生
肌组织的再生能力很弱。横纹肌的再生依肌膜是否存在及肌纤维是否完全断裂而有所不同。横纹肌细胞是一个多核的长细胞,可长达4cm ,核可多达数十乃至数百个,损伤不太重而肌膜未被破坏时,肌原纤维仅部分发生坏死,此时中性粒细胞及巨噬细胞进入该部吞噬清除坏死物质,残存部分肌细胞分裂,产生肌浆,分化出肌原纤维,从而恢复正常横纹肌的结构;如果肌纤维完全断开,断端肌浆增多,也可有肌原纤维的新生,使断端膨大如花蕾样。但这时肌纤维断端不能直接连接,而靠纤维瘢痕愈合。愈合后的肌纤维仍可以收缩,加强锻炼后可以恢复功能;如果整个肌纤维(包括肌膜)均破坏,则难以再生,而通过瘢痕修复。
平滑肌也有一定的分裂再生能力,前面已提到小动脉的再生中就有平滑肌的再生,但是断开的肠管或是较大血管经手术吻合后,断处的平滑肌主要通过纤维瘢痕连接。
心肌再生能力极弱,破坏后一般都是瘢痕修复。
(六)神经组织的再生
脑及脊髓内的神经细胞破坏后不能再生,由神经胶质细胞及其纤维修补,形成胶质瘢痕。外周神经受损时,如果与其相连的神经细胞仍然存活,则可完全再生。首先,断处远侧段的神经纤维髓鞘及轴突崩解,并被吸收;近侧段的数个Ranvier节神经纤维也发生同样变化。然后由两端的神经鞘细胞增生,形成带状的合体细胞,将断端连接。近端轴突以每天约1mm的速度逐渐向远端生长,穿过神经鞘细胞带,最后达到末梢鞘细胞,鞘细胞产生髓磷脂将轴索包绕形成髓鞘(图2-3)。此再生过程常需数月以上才能完成。若断离的两端相隔太远(超过2.5cm时),或者两端之间有瘢痕或其它组织阻隔,或者因截肢失去远端,再生轴突均不能达到远端,而与增生的结缔组织混合在一起,卷曲成团,成为创伤性神经瘤(截肢神经瘤),可发生顽固性疼痛。为防止上述情况发生,临床常施行神经吻合术或对截肢神经断端作适当处理。
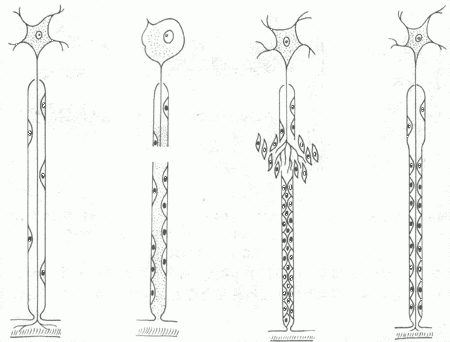
图2-3 神经纤维再生模式图
①正常神经纤维 ②神经纤维断离,远端及近端的一部分髓鞘及轴突崩解③神经膜细胞增生,轴突生长 ④神经轴突达末梢,多余部分消失
三、再生的调控
就单个细胞而言,细胞增殖是受基因控制的,细胞周期出现的一系列变化是基因活化与表达的结果,已知的有关基因包括癌基因(oncogene)及细胞分裂周期基因(cell division cycle gene)。然而机体是由多细胞组成的极其复杂的统一体。部分细胞、组织丧失引起细胞再生予以修复,修复完后成再生便停止,可见机体存在着刺激再生与抑制再生两种机制,两者处于动态平衡。刺激再生的机制增强或抑制再生的机制减弱,则促进再生,否则再生受抑。目前已知短距离调控细胞再生的重要因素包括以下三方面。
1.细胞与细胞之间的作用细胞在生长过程中,如果细胞相互接触,则生长停止,这种现象称为生长的接触抑制。细胞间的缝隙连接(可能还有桥粒)也许参与了接触抑制的调控。肿瘤细胞丧失了接触抑制特性。
2.细胞外基质对细胞增殖的作用实验证明,正常细胞只有粘着于适当的基质才能生长,脱离了基质则很快停止于G1或G0期。基质各种成分对不同细胞的增殖有不同的作用,如层粘连蛋白可促进上皮细胞增殖,抑制纤维母细胞的增殖,而纤维粘连蛋白的作用则正好相反。组织中层粘连蛋白与纤维粘连蛋白的相对比值可能对维持上皮细胞与间质细胞之间的平衡有一定的作用。
3.生长因子及生长抑素的作用近年来分离出许多因子,乃某些细胞分泌的多肽类物质,能特异性地与某些细胞膜上的受体结合,激活细胞内某些酶,引起一系列的连锁反应,从而调节细胞生长、分化。能刺激细胞增殖的多肽称为生长因子(cell growth factors),能抑制细胞增殖的则称为抑素(chalon)。
目前已分离、纯化出一些重要的生长因子,如①表皮生长因子(epidermal growth factor,EGF),对上皮细胞、纤维母细胞、胶质细胞及平滑肌细胞都有促进增殖的作用;②血小板源性生长因子(platelet derived growth factor,PDGF),来源于血小板α颗粒,在凝血过程中释放,对纤维母细胞、平滑肌细胞及胶质细胞的增生有促进作用;③纤维母细胞生长因子(fibroblast growth FGF),能促进多种间质细胞增生及小血管再生;④转化生长因子(transforming growth factor,TGF),最初从肉瘤病毒转化的细胞培养基中分离出来,故名。其实许多正常细胞都分泌TGF。TGF-α与EGF在氨基酸序列方面有33%~44%同源,也可与EGF受体结合,故有相同作用。TGF-β能刺激间质细胞增生;⑤许多细胞因子(cytokines)也是生长因子,例如白介素Ⅰ(IL-1)和肿瘤坏死因子(TNF)能刺激纤维母细胞的增殖及胶原合成,TNF还能刺激血管再生。此外还有许多生长因子,如造血细胞集落刺激因子、神经生长因子、IL-2(T细胞生长因子)等,在此不赘述。
与生子因子相比,对抑素的了解甚少,至今还没有一个抑素被纯化和鉴定。抑素具有组织特异性,似乎任何组织都可产生一种抑素抑制本身的增殖。例如已分化的表皮细胞能分泌表皮抑素,抑制基底细胞增殖。当皮肤受损使已分化的表皮细胞丧失时,抑素分泌中止,基底细胞分裂增生,直到增生分化的细胞达到足够数量和抑素达到足够浓度为止。前面提到的TGF-β虽然对某些间质细胞增殖起促进作用,但对上皮细胞则是一种抑素。此外干扰素-α,前列腺素E2和肝素在组织培养中对纤维母细胞及平滑肌细胞的增生都有抑制作用。
第二节 纤维性修复
纤维性修复首先通过肉芽组织增生,溶解、吸收损伤局部的坏死组织及其它异物,并填补组织缺损,以后肉芽组织转化成以胶原纤维为主的瘢痕组织,这种修复便告完成。
一、肉芽组织
肉芽组织(granulation tissue)乃由旺盛增生的毛细血管及纤维结缔组织和各种炎性细胞组成,肉眼表现为鲜红色,颗粒状,柔软湿润,形似鲜嫩的肉芽故名。
镜下可见大量由内皮细胞增生形成的实性细胞索及扩张的毛细血管,向创面垂直生长,并以小动脉为轴心,在周围形成袢状弯曲的毛细血管网。在毛细血管周围有许多新生的纤维母细胞,此外常有大量渗出液及炎性细胞(图2-4)。炎性细胞中常以巨噬细胞为主,也有多少不等的中性粒细胞及淋巴细胞,因此肉芽组织具有抗感染功能。巨噬细胞能分泌PDGF、FGF、TGF-β、IL-1及TNF,加上创面凝血时血小板释放的PDGF,进一步刺激纤维母细胞及毛细血管增生。巨噬细胞及中性粒细胞能吞噬细菌及组织碎片,这些细胞破坏后释放出各种蛋白水解酶,能分解坏死组织及纤维蛋白,肉芽组织中毛细血管内皮细胞亦有吞噬能力,并有强的纤维蛋白溶解作用。
肉芽组织中一些纤维母细胞的胞浆中含有肌细丝,有收缩功能,因此应称为肌纤维母细胞(myofibroblast)。纤维母细胞产生基质及胶原。早期基质较多,以后则胶原越来越多。
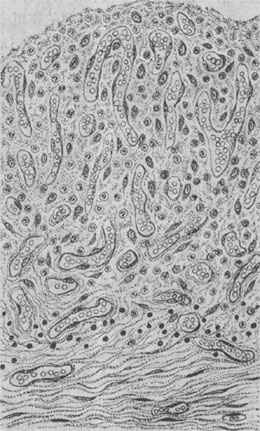
图2-4 肉芽组织镜下结构模式图
图示新生毛细血管、纤维母细胞及各种炎性细胞
1.胶原纤维的生成及分解纤维母细胞在PDGF、FGF、IL-1及TNF等刺激下合成由三股α-肽链互相扭结呈螺旋状的前胶原(procollagen),前胶原分泌到细胞外后,有的被内切酶切去两端的球形结构而成为原胶原(tropocollagen),相邻的原胶原分子互相错开1/4平行排列交联成胶原原纤维(collagenous fibril),在电镜下呈现64nm周期性横纹。胶原原纤维再聚合则成较宽的胶原纤维(collagenous fiber)。从化学成分不同可将胶原分为15种,间质中的胶原纤维主要由Ⅰ、Ⅲ型胶原组成。网状纤维是由Ⅲ型胶原组成的胶原原纤维,由于其纤维表面粘附有较多的蛋白多糖,故染色具有嗜银性,PAS反应阳性。
胶原一般十分稳定,在生理条件下转换率很慢,半生期为数周至数年不等。在病理条件下及胚胎时期转换加快。胶原转换是组织改建所必需的,首先发生胶原的降解,然后由新的胶原形成另外的结构,或者被另一型胶原所取代。胶原对一般蛋白水解酶的抵抗力很强,先要由胶原酶将胶原分子切断,才能由一般的蛋白水解酶降解。胶原酶可由纤维母细胞、巨噬细胞、中性粒细胞及上皮细胞等产生,经某些蛋白酶激活后才能具有活性。不同来源的胶原酶对不同类型胶原的降解能力不同。
2.细胞外基质主要有两大类:①粘连蛋白如纤维粘连蛋白(fibronectin)及层粘连蛋白(laminin)。纤维粘连蛋白除纤维母细胞外,内皮细胞、巨噬细胞及许多上皮细胞均可合成。间质中的纤维粘连蛋白在基质各成分之间及与细胞之间起连接作用。层粘连蛋白存在于基底膜中,由基底膜上的细胞如上皮细胞、内皮细胞等合成。如前所述,这两种粘连蛋白对细胞的生长与分化有调控作用;②氨基多糖与蛋白多糖。氨基多糖(glycosaminglycan)亦称酸性粘多糖,包括透明质酸、硫酸软骨素、硫酸皮肤素、硫酸角质素、硫酸乙酰肝素等,在肉芽组织中主要是透明质酸及硫酸软骨素。除透明质酸外,其他氨基多糖能与核心蛋白结合而形成蛋白多糖(proteoglycan),以前称粘蛋白。氨基多糖及蛋白多糖组成多孔亲水的凝胶结构,有利于水分及小分子的渗透,有的对细胞的生长、分化有胶原形成还有调节作用。它们由纤维母细胞及类似细胞(如骨母细胞、软骨母细胞)合成,一些多糖酶如透明质酸酶可将其降解。
肉芽组织在修复过程中的作用:①机化血凝块、坏死组织及其他异物;②抗感染及保护创面;③填补伤口及其它组织缺损。
二、瘢痕组织
瘢痕组织(scar tissue)的形成是肉芽组织逐渐纤维化的过程。此时网状纤维及胶原纤维越来越多,网状纤维胶原化,胶原纤维变粗,与此同时纤维母细胞越来越少,少量剩下者转变为纤维细胞;间质中液体逐渐被吸收,中性粒细胞、巨噬细胞、淋巴细胞和浆细胞先后消失;毛细血管闭合、退化、消失,留下很少的小动脉及小静脉。这样,肉芽组织乃转变成主要由胶原纤维组成的血管稀少的瘢痕组织,肉眼呈白色,质地坚韧。
瘢痕形成宣告修复完成,然而瘢痕本身仍在缓慢变化:如常发生玻璃样变,有的瘢痕则发生瘢痕收缩,这种现象不同于创口的早期收缩,而是瘢痕在后期由于水分的显著减少所引起的体积变小,有人认为也与肌纤维母细胞持续增生以至瘢痕中有过多的肌纤维母细胞有关。由于瘢痕坚韧又缺乏弹性,加上瘢痕收缩可引起器官变形及功能障碍,如在消化道、泌尿道等腔室器官则引起管腔狭窄,在关节附近则引起运动障碍;一般情况下,瘢痕中的胶原还会逐渐被分解、吸收,以至改建,因此瘢痕会缓慢地变小变软;但偶尔也有的瘢痕胶原形成过多,成为大而不规则的隆起硬块,称为瘢痕疙瘩(keloid),易见于烧伤或反复受异物等刺激的伤口,其发生机制不明,一般认为与体质有关。那些容易出现瘢痕疙瘩的人的体质称为瘢痕体质。瘢痕疙瘩中的血管周围常见一些肥大细胞,故有人认为,由于持续局部炎症及低氧,促进肥大细胞分泌多种生长因子,使肉芽组织过度生长,因而形成瘢痕疙瘩。
第三节 创伤愈合
创伤愈合(wound healing)是指机体遭受外力作用,皮肤等组织出现离断或缺损后的愈复过程,为包括各种组织的再生和肉芽组织增生、瘢痕形成的复杂组合,表现出各种过程的协同作用。
一、创伤愈合的基本过程
最轻度的创伤仅限于皮肤表皮层,稍重者有皮肤和皮下组织断裂,并出现伤口;严重的创伤可有肌肉、肌腱、神经的断裂及骨折。下述有伤口的创伤愈合的基本过程。
1.伤口的早期变化伤口局部有不同程度的组织坏死和血管断裂出血,数小时内便出现炎症反应,表现为充血、浆液渗出及白细胞游击,故局部红肿。白细胞以中性粒细胞为主,3天后转为以巨噬细胞为主。伤口中的血液和渗出液中的纤维蛋白原很快凝固形成凝块,有的凝块表面干燥形成痂皮,凝块及痂皮起着保护伤口的作用。
2.伤口收缩2~3天后伤口边缘的整层皮肤及皮下组织向中心移动,于是伤口迅速缩小,直到14天左右停止。伤口收缩的意义在于缩小创面。实验证明,伤口甚至可缩小80%,不过在各种具体情况下伤口缩小的程度因动物种类、伤口部位、伤口大小及形状而不同。伤口收缩是伤口边缘新生的肌纤维母细胞的牵拉作用引起的,而与胶原无关。因为伤口收缩的时间正好是肌纤维母细胞增生的时间。5-HT、血管紧张素及去甲肾上腺素能促进伤口收缩,糖皮质激素及平滑肌拮抗药则能抑制伤口收缩。抑制胶原形成则对伤口收缩没有影响,植皮可使伤口收缩停止。
3.肉芽组织增生和瘢痕形成大约从第3天开始从伤口底部及边缘长出肉芽组织,填平伤口。毛细血管大约以每日延长0.1~0.6mm的速度增长,其方向大都垂直于创面,并呈袢状弯曲。肉芽组织中没有神经,故无感觉。第5~6天起纤维母细胞产生胶原纤维,其后一周胶原纤维形成甚为活跃,以后逐渐缓慢下来。随着胶原纤维越来越多,出现瘢痕形成过程,大约在伤后一个月瘢痕完全形成。可能由于局部张力的作用,瘢痕中的胶原纤维最终与皮肤表面平行。
瘢痕可使创缘比较牢固地结合。伤口局部抗拉力的强度于伤后不久就开始增加,在第3~5周抗拉力强度增加迅速,然后缓慢下来,至3个月左右抗拉力强度达到顶点不再增加。但这时仍然只达到正常皮肤强度的70%~80%。伤口抗拉力的强度可能主要由胶原纤维的量及其排列状态决定,此外,还与一些其它组织成分有关。腹壁切口愈合后,如果瘢痕形成薄弱,抗拉强度较低,加之瘢痕组织本身缺乏弹性,故腹腔内压的作用有时可使愈合口逐渐向外膨出,形成腹壁疝。类似情况还见于心肌及动脉壁较大的瘢痕处,可形成室壁瘤及动脉瘤。
表皮及其它组织再生 创伤发生24小时以内,伤口边缘的表皮基底增生,并在凝块下面向伤口中心移动,形成单层上皮,覆盖于肉芽组织的表面,当这些细胞彼此相遇时,则停止前进,并增生、分化成为鳞状上皮。健康的肉芽组织对表皮再生十分重要,因为它可提供上皮再生所需的营养及生长因子,如果肉芽组织长时间不能将伤口填平,并形成瘢痕,则上皮再生将延缓;在另一种情况下,由于异物及感染等刺激而过度生长的肉芽组织(exuberant granulation),高出于皮肤表面,也会阻止表皮再生,因此临床常需将其切除。若伤口过大(一般认为直径超过20cm时),则再生表皮很难将伤口完全覆盖,往往需要植皮。
皮肤附属器(毛囊、汗腺及皮脂腺)如遭完全破坏,则不能完全再生,而出现瘢痕修复。肌腱断裂后,初期也是瘢痕修复,但随着功能锻炼而不断改建,胶原纤维可按原来肌腱纤维方向排列,达到完全再生。
二、创伤愈合的类型
根据损伤程度及有无感染,创伤愈合可分为以下三种类型。
1.一般愈合(healing by first intention)见于组织缺损少、创缘整齐、无感染、经粘合或缝合后创面对合严密的伤口,例如手术切口。这种伤口中只有少量血凝块,炎症反应轻微,表皮再生在24~48小时内便可将伤口覆盖。肉芽组织在第三天就可从伤口边缘长出并很快将伤口填满,5~6天胶原纤维形成(此时可以拆线),约2~3周完全愈合,留下一条线状瘢痕。一期愈合的时间短,形成瘢痕少(图2-5)。
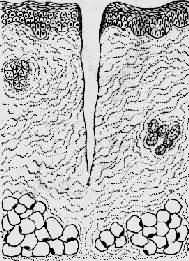
1.创缘整齐,组织破坏少
2.经缝合,创缘对合,炎症反应轻
3.表皮再生,少量肉芽组织从伤口缘长入
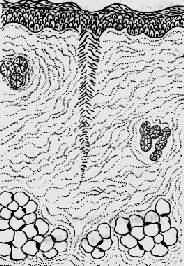
4.愈合后少量疤痕形成
图2-5 创伤一期愈合模式图
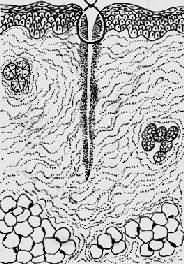
2.二期愈合(healing by second intention) 见于组织缺损较大、创缘不整、哆开、无法整齐对合,或伴有感染的伤口。这种伤口的愈合与一期愈合有以下不同:①由于坏死组织多,或由于感染,继续引起局部组织变性、坏死,炎症反应明显。只有等到感染被控制,坏死组织被清除以后,再生才能开始。②伤口大,伤口收缩明显,从伤口底部及边缘长出多量的肉芽组织将伤口填平。③愈合的时间较长,形成的瘢痕较大(图2-6)。
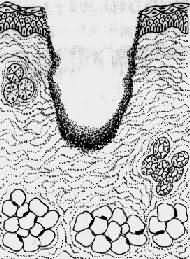
1.创口大,创缘不整,组织破坏多
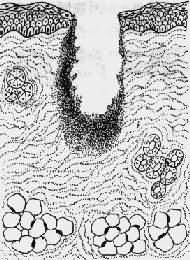
2.伤口收缩,炎症反应重
3.肉芽组织从伤口底部及边缘将伤口填平,然后表皮再生

4.愈合后形成疤痕大
图2-6 创伤二期愈合模式图
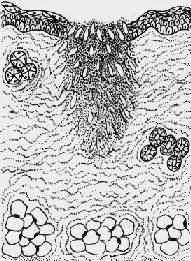
3.痂下愈合(healing under scab) 伤口表面的血液、渗出液及坏死物质干燥后形成黑褐色硬痂,在痂下进行上述愈合过程。待上皮再生完成后,痂皮即脱落。痂下愈合所需时间通常较无痂者长,因此时的表皮再生必须首先将痂皮溶解,然后才能向前生长。痂皮由于干燥不利于细菌生长,故对伤口有一定的保护作用。但如果痂下渗出物较多,尤其是已有细菌感染时,痂皮反而成了渗出物引流排出的障碍,使感染加重,不利于愈合。
三、影响再生修复的因素
从上述可以看出,损伤的程度及组织的再生能力决定修复的方式、愈合的时间及瘢痕的大不。因此,治疗原则应是缩小创面(如对合伤口)、防止再损伤和促进组织再生。虽然组织的再生能力是在进化过程中获得的,但仍受全身及局部条件的影响。因此,应当避免一些不利因素,创造有利条件促进组织再生修复。此外,由于瘢痕组织在一定条件下可以造成危害,因而有时需要抑制瘢痕的形成或者促进瘢痕的吸收。
影响再生修复的因素包括全身因素及局部因素两方面。
(一)全身因素
1.年龄 青少年的组织再生能力强,愈合快。老年人则相反,组织再生能力差,愈合慢,与老年人血管硬化、血液供应减少有很大的关系。
2.营养 严重的蛋白质缺乏,尤其是含硫氨基酸(如甲硫氨酸、胱氨酸)缺乏时,肉芽组织及胶原形成不良,伤口愈合延缓。维生素中以维生素C对愈合最重要。这是由于α-多肽链中的两个主要氨基酸—脯氨酸及赖氨酸,必须经羟化酶羟化,才能形成前胶原分子,而维生素C具有催化羟化酶的作用,因此维生素C缺乏时前胶原分子难以形成,从而影响了胶原纤维的形成。在微量元素中锌对创伤愈合有重要作用,手术后伤口愈合迟缓的病人,皮肤中锌的含量大多比愈合良好的病人低。此外已证明,手术刺激、外伤及烧伤患者尿中锌的排出量增加,补给锌能促进愈合。锌的作用机制不很清楚,可能与锌是细胞内一些氧化酶的成分有关。
(二)局部因素
1.感染与异物感染对再生修复的妨碍甚大。许多化脓菌产生一些毒素和酶,能引起组织坏死,基质或胶原纤维溶解。这不仅加重局部组织损伤,也防碍愈合。伤口感染时,渗出物很多,可增加局部伤口的张力,常使正在愈合的伤口或已缝合的伤口裂开,或者导致感染扩散加重损伤。因此,对于感染的伤口,不能缝合,应及早引流,只有感染被控制后,修复才能进行。此外,坏死组织及其它异物,也妨碍愈合并有利于感染。因此,伤口如有感染,或有较多的坏死组织及异物,必然是二期愈合。临床上对于创面较大、已被细菌污染但尚未发生明显感染的伤口,施行清创术以清除坏死组织,以缩小创面。这样,可以使本来应是二期愈合的伤口,愈合的时间缩短,甚至可能达到一期愈合。
2.局部血液循坏 局部血液循环一方面保证组织再生所需的氧和营养,另一方面对坏死物质的吸收及控制局部感染也起重要作用。因此,局部血流供应良好时,则再生修复好,相反,如下肢血管有动脉粥样硬化或静脉曲张等病变,使局部血液循环不良时,则该处伤口愈合迟缓。临床用某些药物湿敷、热敷以及帖敷中药和服用活血化瘀中药等,都有改善局部血液循环的作用。
3.神经支配 完整的神经支配对组织再生有一定的作用。例如麻风引起的溃疡不易愈合,是因为神经受累的缘故。植物神经的损伤,使局部血液供应发生变化,对再生的影响更为明显。
4.电离辐射 能破坏细胞,损伤小血管,抑制组织再生。因此能阻止瘢痕形成。
第三章 局部血液及体液循环障碍
血液和体液的局部循环障碍,包括①局部循环血量的异常,包括充血和缺血;②血液性状和血管内容物的异常,包括血栓形成、栓塞及梗死;③水肿和积液。
第一节 充血
器官或局部组织的血管内血液含量增多称为充血(hyperemia),分为动脉性充血和静脉性充血两类。
一、动脉性充血
局部器官或组织由于动脉血输入量增多而发生的充血,称为动脉性充血(arteial hyperemia),又称主动性充血(active hyperemia),简称充血。
【原因】
凡能引起细动脉扩张的任何原因,都可引起局部器官和组织的充血。
细动脉扩张是神经体液因素作用于血管,使血管舒张神经兴奋性增高或血管收缩神经兴奋性降低的结果。许多动脉性充血是器官和组织的正常的生理性活动,如进食后的胃肠粘膜充血,体力运动时横纹肌充血等。这种由于生理性代谢增强所引起的局部充血,称为生理性充血。有些病理过程的某一阶段,也有动脉性充血的参与,例如炎症反应的初始,由于致炎因子的刺激所导致的轴索反射和组胺等血管活性物质的作用,局部组织的细动脉扩张,这时的充血,是动脉性充血。此外,体内还有不少反应性的动脉性充血,例如局部器官和组织长期受压(如绷带包扎肢体或腹水压迫腹腔器官)后,组织内的血管张力降低,若一旦压力突然解除,爱压组织内的细动脉乃发生反射性扩张,发生局部充血,称为减压后充血。
【病变及后果】
动脉性充血的器官和组织内血量增多,体积可轻度增大。充血如发生于体表,可见局部组织的颜色鲜红,温度升高。动脉性充血是暂时性的血管反应,原因消除后,局部血量迅即恢复正常,不遗留不良后果,对机体无重要影响。炎症反应的动脉性充血,是一系列血管反应的初始,它参与炎症血管现象,具有积极的作用。
二、静脉性充血
局部器官或组织由于静脉血液回流受阻使血液淤积于小静脉和毛细血管内而发生的充血,称为静脉性充血(venous hyperemia),又称被动性充血(passive hyperemia),简称淤血(congestion)。
静脉性充血远较动脉性充血多见,具有重要的临床和病理意义。它可发生于局部,也可发生于全身。
【原因】
静脉性充血的原因很多,可归纳为以下三类:
1.静脉受压静脉受压使其管腔发生狭窄或闭塞,血液回流受阻可导致相应部位的器官和组织发生静脉性充血。常见的例子如妊娠子宫压迫髂静脉引起的下肢静脉性充血,肿瘤压迫静脉引起相应器官或组织的静脉性充血,肠套叠、肠扭转和肠疝时肠系膜静脉受压引起局部肠段严重的静脉性充血,以及肝硬变时肝内肝静脉分支受增生肝实质结节压迫引起门静脉所属器官的静脉性充血等。
2.静脉腔阻塞静脉血栓形成可以造成静脉腔的阻塞,可以引起相应器官或组织的静脉性充血。但由于静脉的分支多,只有当静脉腔阻塞而血流又不能充分地通过侧支回流时,才发生静脉性充血。
3.心力衰竭二尖瓣瓣膜病和高血压病引起左心衰竭时,可导致肺淤血;肺源性心脏病引起的右心衰竭,可导致大循环淤血。
【病变】
静脉性充血的组织和器官,可由于血液的淤积而肿胀;发生于体表的静脉性充血,由于血液内氧合血红蛋白减少,还原血红蛋白增多,局部可呈紫绀;又由于局部血流淤滞,毛细血管扩张,使得散热增加,该处体表的温度因而降低。静脉性充血的组织,镜下呈小静脉和毛细血管扩张,充满血液,有时还伴有水肿。由于局部血液氧分压降低,器官和组织相对缺氧,代谢功能可因而减弱。
【后果】
静脉性充血对机体的影响决定于淤血的范围、淤血的器官、淤血的程度、淤血发生的速度(急性或慢性)以及侧支循环建立的状况。全身性淤血影响许多重要器官的功能,可出现相应的功能障碍(如肾、肝、肺),局部性静脉性充血则主要影响局部器官的功能。
较长期的静脉性充血,使局部组织内代谢中间产物蓄积,从而损害毛细血管,使其通透性增高,加之淤血时小静脉和毛细血管内流体静力压升高,导致局部组织发生水肿,严重时甚至发生漏出性出血。如肺淤血时,肺泡壁毛细血管扩张、充血,严重时肺泡腔内可出现水肿液,甚至出血。若肺泡腔内的红细胞被巨噬细胞吞噬,其血红蛋白变为含铁血黄素,使痰呈褐色。这种巨噬细胞常在左心衰竭的情况下出现,因而被称为心力衰竭细胞(heart failure cell)(图3-1)。
长期淤血,由于氧和营养物质供应不足和代谢中间产物堆积,还可引起实质细胞的萎缩和变性。如慢性肝淤血时(图3-2),肝细胞萎缩(主要在肝小叶中央带)和脂肪变(主要在小叶周边带),以致肝切面呈现槟榔状花纹,称为槟榔肝(nutmeg liver)。较急性且程度严重的肝淤血可引起肝细胞坏死。
某些器官,慢性淤血引起实质细胞萎缩的同时,其间质细胞却可增生。例如慢性肝淤血时,小叶中央肝细胞萎缩,结缔组织则增生,最后形成淤血性肝硬变。
由于静脉通常都有丰富的吻合支,因此当某一静脉发生阻塞时,其吻合支能及时扩张,有助于局部血液回流,起了代偿作用。这种通过吻合支的血液流通,称为侧支循环。最典型的例子是肝硬变时部分门静脉系统的静脉血流通过侧支循环回纳于右心。重要的途径如下:①经胃冠状静脉→食管下静脉→奇静脉→上腔静脉→;此时食管下段和胃底的粘膜静脉高度扩张、弯曲,突出于粘膜表面;②经重新开放的脐静脉→脐周腹壁静脉→腹上静脉→乳内静脉→上腔静脉,患者脐周腹壁静脉高度扩张、弯曲,形成所谓“海蛇头”(caput medusae)样的形态;③经肠系膜下静脉→痔静脉丛→髂内静脉→下腔静脉,痔静脉丛高度扩张,形成痔疮。
侧支循环具有一定程度的代偿作用,但当淤血的程度超过侧支循环所能代偿的范围时,终归会出现静脉性充血所招致的各种病理变化。例如肝硬变时尽管有上述的侧支循环,患者仍然出现淤血性脾肿大、胃肠淤血和腹水。此外,侧支循环虽然有代偿静脉回流的积极意义,但侧支静脉过度曲张,有时却可继发地发生破裂(例如食管下段的静脉曲张,可发生致命的大出血)。
图3-1 慢性肺淤血
肺泡壁毛细血管充血,肺泡腔内漏出性出血并出现心力衰竭细胞

图3-2 慢性肝淤血
肝窦扩张充血,肝细胞脂肪变性
第二节 出血
血液自心、血管腔外出,称为出血(hemorrhage)。流出的血液逸入体腔或组织内者,称为内出血,血液流出体外称为外出血。
按血液逸出的机制可将出血分为破裂性出血和漏出性出血两种:
(一)破裂性出血
破裂性出血乃由心脏或血管壁破裂所致。破裂可发生于心脏(如心壁瘤的破裂),也可发生于动脉,其成因既可为动脉壁本身的病变(如主动脉瘤),也可因动脉旁病变侵蚀动脉壁(如肺结核空洞对肺血管壁的破坏,肺癌、胃癌、子宫颈癌的癌组织侵蚀局部血管壁,胃和十二指肠慢性溃疡的溃疡底的血管被病变侵蚀)。静脉破裂性出血的原因除创伤外,较常见的例子是肝硬变时食管静脉曲张的破裂。毛细血管的破裂性出血发生于局部软组织的损伤。
(二)漏出性出血
这种出血是由于毛细血管后静脉、毛细血管以及毛细血管前动脉的血管壁通透性增高,血液通过扩大的内皮细胞间隙和受损的血管基底膜而漏出于管腔外的。出血性素质所发生的自发性出血,即是漏出性出血。
【原因】
漏出性出血的原因很多,基本可归纳为:
1.血管壁损害常见于缺氧,使毛细血管内皮细胞变性;败血症(尤其是脑膜炎球菌败血症)、立克次体感染、流行性出血热、蛇毒、有机磷中毒等使毛细血管壁损伤;一些药物可引起变态反应性血管炎;维生素C缺乏可引起毛细血管基底膜破裂、毛细血管周胶原减少及内皮细胞连接处分开而致管壁通透性升高;过敏性紫癜时由于免疫复合物沉着于血管壁引起变态反应性血管炎。
2.血小板减少和功能障碍血小板的正常数量和质量是维持毛细血管通透性正常的重要因素,血小板减少到一定数量时即可发生漏出性出血,例如再生障碍性贫血、白血病、骨髓内广泛性肿瘤转移等均可使血小板生成减少;原发性血小板减少性紫癜、血栓性血小板减少性紫癜、DIC使血小板破坏或消耗过多;某些药物在体内诱发抗原抗体复合物免疫反应所形成的免疫复合物吸附于血小板表面,使后者连同免疫复合物被巨噬细胞所吞噬;一些细菌的内毒素和外毒素也有破坏血小板的作用。
血小板的结构和功能缺陷也能引起漏出性出血,这类疾患很多是先天性的,如血小板功能不全(thrombasthenia,血小板细胞膜缺乏纤维蛋白受体)和血小板颗粒缺乏症(storage pool disease,一种或多种颗粒缺乏,ADP储量因而不足;也可因后天性骨髓巨核细胞受损而发生)时,血小板粘集能力有缺陷;Bernard-Soulier综合征(血小板细胞膜缺乏von Willebrand因子的受体)时,血小板不能粘附于胶原纤维,这都可有凝血障碍或出血倾向。
3.凝血因子缺乏 凝血因子Ⅷ(血友病A)、Ⅸ(血友病B)、von Willebrand因子(von Willebrand病)以及纤维蛋白原、凝血酶原、Ⅳ、Ⅴ、Ⅶ、Ⅹ、Ⅺ等因子的先天性缺乏或肝实质疾患时凝血因子Ⅶ、Ⅸ、Ⅹ合成减少,DIC时凝血因子消耗过多)等,均有出血倾向。
【病变】
内出血可发生于体内任何部位,血液积聚于体腔内者称体腔积血,如腹腔积血、心包积血;体腔内可见血液或凝血块。发生于组织内的出血,量大时形成血肿(hematoma),如脑血肿、皮下血肿等;量少时仅镜下始能查觉,在组织内有多少不等的红细胞或含铁血黄素、橙色血晶(hematoidin)的存在。皮肤、粘膜、浆膜的少量出血在局部形成瘀点(petechia),较大的出血灶形成瘀斑(echymosis)。
【后果】
出血对机体的影响取决于出血量、出血速度和出血部位。漏出性出血过程比较缓慢,出血量较少,不会引起严重后果。但如漏出性出血广泛时,如肝硬变时因门静脉高压发生的广泛性胃肠粘膜漏出性出血,可因一时的多量出血导致出血性休克。破裂性出血的出血过程迅速,如在短时间内丧失循环血量的20%~25%时,即可发生出血性休克。发生在重要器官的出血,即使出血量不多,亦可致命,如心脏破裂引起心包内出血,由于心包填塞,可导致急性心功能不全;脑出血,尤其是脑干出血,可因重要神经中枢受压致死。局部的出血,可导致相应的功能障碍,如脑内囊出血引起对侧肢体偏瘫,视网膜出血引起视力减退或失明。慢性出血可引起贫血。
一般的进行缓慢的破裂性出血,多可自行停止。其机制是局部受损的细动脉发生痉挛,小静脉形成血栓,从而阻止血液继续流失。流入体腔或组织内的血液,久后可被吸收、机化或包裹。
第三节 血栓形成
在活体的心脏或血管腔内,血液发生凝固或血液中的某些有形成分互相粘集,形成固体质块的过程,称为血栓形成(thrombosis),在这个过程中所形成的固体质块称为血栓(thrombus)。
血液中存在着相互拮抗的凝血系统和抗凝血系统(纤维蛋白溶解系统)。在生理状态下,血液中的凝血因子不断地被激活,从而产生凝血酶,形成微量纤维蛋白,沉着于血管内膜上,但这些微量的纤维蛋白又不断地被激活了的纤维蛋白溶解系统所溶解,同时被激活的凝血因子也不断地被单核吞噬细胞系统所吞噬。上述凝血系统和纤维蛋白溶解系统的动态平衡,即保证了血液有潜在的可凝固性又始终保证了血液的流体状态。然而,有时在某些能促进凝血过程的因素作用下,打破了上述动态平衡,触发了凝血过程,血液便可在心血管腔内凝固,形成血栓。
一、血栓形成的条件和机制
凝血系统在流动的血液中被激活,必须具备一定的条件:
1.心血管内膜的损伤内皮细胞有一系列的防止血液在心血管内凝固的功能:①它是一个单细胞层的薄膜屏障,把血液中的凝血因子、血小板和能促发凝血的内皮下胶原隔离开来;②能分泌凝血酶调节蛋白(thrombomodulin),它是一种糖蛋白覆于内膜表面,能和凝血酶结合而控制凝血酶的作用;③合成前列环素,能抑制血小板粘集;④分泌二磷酸腺苷酶,把血小板释出的、对血小板彼此粘集具有强烈作用的ADP转变为抗粘集作用的腺嘌呤核苷酸;⑤内皮细胞的细胞膜表面含有肝素样分子(硫酸乙酰肝素),它有促进抗凝血酶Ⅲ的作用;又含有α2巨球蛋白,它能抑制凝血因子活化过程的链锁反应。⑥生成纤溶酶原活化因子,有促进纤维蛋白溶解的作用。⑦合成蛋白S(PS),它能协同活化的蛋白C(PC)抑制凝血因子Ⅴa、Ⅷa(a表示活化的)。由于以上因素,如果没有心血管内膜的损伤,超出生理限度的血小板和凝血因子的活化就不可能发生。况且,内皮细胞还具有促血凝的作用,因它能合成组织因子,存于内皮细胞内,内皮细胞损伤时得以释出,从而激活外途径凝血过程。
内皮细胞损伤暴露出皮下的胶原,这对活化血小板和凝血因子Ⅻ至关重要。内皮下结缔组织内的纤维连接蛋白也有助于血液细胞和纤维蛋白原粘着在暴露的血管壁上。此外,内皮下由血小板和内皮细胞所生成的凝血酶敏感蛋白(thrombospondin,一种糖蛋白)也可和纤维蛋白原和纤维连接蛋白等大分子结合,使血细胞和血管壁粘连。然而在触发凝血过程中起核心作用的是血小板的活化。能激活血小板的物质有胶原、凝血酶、ADP和凝血恶烷A2(thromboxane A2)等,在内皮损伤后,首先激活血小板的是与血小板接触的胶原,继后凝血链锁反应被启动而产生了凝血酶,并且血小板继续地被活化又不断释出ADP和血栓素A2,随血流而来的是血小板在局部不断地被激活。血小板活表现为下述三项反应:①粘附反应,血小板粘附于局部胶原(需要有内皮细胞所合成的von Willebrand因子的介入),同时由于其胞浆内微丝和微管的收缩而变形,血小板的是颗粒逐渐消失而使胞浆同质化。②释放反应,血小板的α颗粒(含有纤维蛋白原、纤维连接蛋白、抗肝素即血小板第4因子、血小板生长因子及血小板所合成的凝血酶敏感白)和致密颗粒(含有丰富的ADP、Ca离子、去甲肾上腺素、组胺、5-HT)的内容物向血小板外释出,其中ADP对在此经过的血中血小板不断地互相粘集起了重大作用。与此同时,位于血小板膜的第3因子(磷脂)也暴露于细胞膜,成为和凝血因子Ⅸa、Ⅷa、Ca2+结合的场所,X在这里被激活后,Xa、Va、Ca2+也在这里结合,形成凝血酶原酶、将凝血酶原激活成凝血酶。③粘集反应,促使血小板彼此粘集成集群的因子主要是ADP、血栓素A2和凝血酶。最起始的粘集是通过释放反应所释出的ADP,在ADP量少的情况下,所形成的血小板粘集堆是可复性的,即一旦血流加速,粘集成堆的血小板仍可一一散开;但随着血小板愈粘集愈多,活化后释出的ADP也愈多,粘集堆逐成为不可复性。促成不可复性粘集的另一因子是血小板活化时所生成的血栓素A2,后者既有强大的促粘集性,又有使血小板发生释放反应的功能。在凝血因子Ⅻ(内途径)和Ⅶ(外途径)分别被胶原和组织因子所激活、凝血反应的产物凝血酶形成后,凝血酶、ADP、血栓素A2共同使血小板粘集堆成为持久性。血栓形成是以在胶原暴露的局部形成持久性血小板粘集堆开始的,因此血栓多见于静脉内膜炎、结节性多动脉炎、动脉粥样硬化溃疡、风湿性和细菌性心内膜炎、心肌梗死等病变的心血管内膜(壁)上。
2.血流状态的改变由于比重的关系,在正常流速和正常流向的血液内,红细胞和白细胞在血流的中轴(轴流),外层是血小板,流动得较红、白细胞缓慢,是外围是一层血浆带(边流),将血液的有形成分和血管壁隔绝,阻止血小板和内膜接触。当血流缓慢或血流产生漩涡时,血小板得以进入边流,增加了和血管内膜接触的机会,血小板粘连于内膜的可能性必然增大。此外,血流缓慢和血流产生漩涡时,被激活的凝血因子和凝血酶能在局部达到凝血过程所必需的浓度。尽管在光学显微镜下,血流缓慢并不造成可以察觉的内膜变化,但电镜下却可发现血流缓慢,严重缺氧时,内皮细胞胞浆出现空泡,最后整个细胞变成无核结构的物质,由此不难推论,内皮细胞的变性坏死,不但丧失了上述的抗凝血因子的合成和分泌,而且内皮下胶原也得以暴露于血流,这样,即可触发内源性和外源性凝血途径。不少事实表明血流缓慢是血栓形成的重要因素,例如静脉发生血栓约比动脉发生血栓多4倍,静脉血栓常发生于久病卧床的患者和静脉曲张的静脉内等。静脉比动脉容易发生血栓,除了血流缓慢因素外,还因静脉有静脉瓣,静脉瓣内的血流不但缓慢,而且呈漩涡,因此静脉血栓形成往往以瓣膜囊为起始点;此外,静脉不似动脉那样随心脏搏动而舒张,其血流有时甚至可出现短暂的停滞;静脉壁较薄,容易受压;血流通过毛细血管到静脉后,血液的粘性有所增加等因素。心脏和动脉内的血流快,不易形成血栓,但在血流较缓和出现漩涡时,也会有血栓形成,如二尖瓣狭窄时左心房血流缓慢并出现漩涡,动脉瘤内的血流呈漩涡状流动,这时均易并发血栓形成。
3.血液凝固性增加血液凝固性增加,或称血液的高凝状态,是指血液比正常易于发生凝固的状态,见于弥散性血管内凝血(DIC)和游走性血栓性脉管炎(Trausseau综合征)。DIC的血液凝固性增加是由于一系列因素所诱发的凝血因子激活,或有组织因子的释出,Trausseau综合征则发生于一些癌肿,尤其是胰腺癌、胃癌、乳腺癌和支气管癌,其血液的凝固性增加系由于癌细胞释出促凝因子,如组织因子、促凝血因子A(procoagulant A)等。此外,血小板增多或血小板粘性增加也可增高血液的凝固性,如妊娠、手术后、产后、高脂饮食、吸烟、冠状动脉粥样硬化时,血栓形成的可能性增加均与此有关。
需要强调的是,上述血栓形成条件,往往是同时存在的。例如手术后卧床、创伤、晚期癌全身转移时的血栓形成,既由于血液的凝固性增加,又由于静卧时血流缓慢和下肢静脉(尤其是腓肠肌内的静脉)受压。
二、血栓形成的过程及血栓的形态
无论心或动脉、静脉内的血栓,其形成过程都从血小板粘附于内膜裸露的胶原开始。当内源性和外源性凝血途径启动后最后产生的凝血酶将纤维蛋白原水解,其纤维蛋白单体再聚合成纤维蛋白多聚体(纤维素)。纤维素和内皮下的纤维连接蛋白共同使粘集的血小板堆牢固地粘附于受损内膜表面,不再离散,形成境下均匀一致、无结构的血小板血栓,电子显微镜下,血小板彼此紧密接触,轮廓仍然保存,但内部颗粒已消失。在血小板与血小板之间有少量纤维素存在(图3-3)。

图3-3 血小板粘附
血管内皮细胞脱落,血小板粘附在暴露的纤维结缔组织上
(电子显微镜照片)(采自Anderson)
血小板粘集堆的形成是血栓形成的第一步,嗣后血栓形成的过程及血栓的组成、形态、大小都取决于血栓发生的部位和局部血流速度(图3-4)。
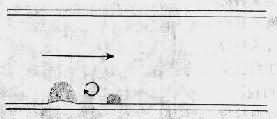
血管内膜粗糙,血小板粘集成堆,使局部血流形成漩涡

血小板继续粘集形成多数小梁,小梁周围有白细胞粘附

小梁间形成纤维素网,网眼中充满红细胞
血管腔阻塞,局部血流停滞,停滞之血液凝固
图3-4 血栓形成过程示意图
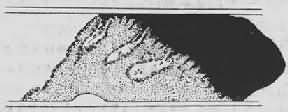
血栓大致可分为以下几种类型:
1.白色血栓(pale thrombus)发生于血流较速的部位(如动脉、心室)或血栓形成时血流较速的时期〔如静脉混合性血栓的起始部,即延续性血栓(propagating thrombus)的头部〕。镜下,白色血栓主要由许多聚集呈珊瑚状的血小板小梁构成,其表面有许多中性白细胞粘附,形成白细胞边层,推测是由于纤维素崩解产物的趋化作用吸引而来。血小板小梁之间由于被激活的凝血因子的作用而形成网状的纤维素,其网眼内含有少量红细胞。肉眼观呈灰白色,表面粗糙有波纹,质硬,与血管壁紧连。
2.红色血栓(red thrombus)发生在血流极度缓慢甚或停止之后,其形成过程与血管外凝血过程相同。因此,红色血栓见于混合血栓逐渐增大阻塞管腔,局部血流停止后,往往构成延续性血栓的尾部。镜下,在纤维素网眼内充满如正常血液分布的血细胞。肉眼观呈暗红色。新鲜的红色血栓湿润,有一定的弹性,陈旧的红色血栓由于水分被吸收,变得干燥,易碎,失去弹性,并易于脱落造成栓塞。
3.混合血栓(mixed thrombus)静脉的延续性血栓的主要部分(体部),呈红色与白色条纹层层相间,即是混合性血栓。其形成过程是:以血小板小梁为主的血栓不断增长以致其下游血流形成漩涡,从而再生成另一个以血小板为主的血栓,在两者之间的血液乃发生凝固,成为以红细胞为主的血栓。如是交替进行,乃成混合性血栓(图3-5)。在二尖瓣狭窄和心房纤维颤动时,在左心房可形成球形血栓;这种血栓和动脉瘤内的血栓均可见到灰白色和红褐色交替的层状结构,称为层状血栓,也是混合性血栓。
4.透明血栓(hyaline thrombus)这种血栓发生于微循环小血管内,只能在显微镜下见到,故又称微血栓,主要由纤维素构成,见于弥散性血管内凝血。
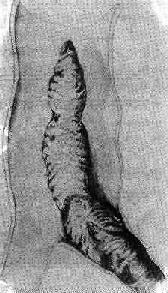
图3-5 延续性血栓
血栓形成于髂静脉内,向下腔静脉延续,当延续到对侧髂静脉入口处后,由于有血流流入,血小板继续析出,继续形成白色血栓,顺血流延伸,并常游离于血管腔内而不与血管壁粘连(采自Eder )
三、血栓的结局
1.软化、溶解、吸收当第Ⅻ因子被激活后,活化的第Ⅻ因子已开始激活纤维蛋白溶酶系统,裂解纤维蛋白原和纤维素,血栓的持续存在并增长,抑或软化、溶解、吸收,取决于凝血系统和纤维蛋白溶酶系统两者之间活性的对比。血栓内的白细胞,其溶蛋白酶也有溶解血栓成分的能力。当溶解血栓成分的酶量多、活性强时,血栓可被溶解,小的血栓可完全被溶解吸收。
2.机化血栓形成后,从血管壁向血栓长入内皮细胞和纤维母细胞,随即形成肉芽组织,血小板的血小板生长因子可能起着促使肉芽组织生长的作用。肉芽组织伸入血栓,逐渐加以取代而发生机化。机化过程早在血栓形成后1~2天即已开始,较大的血栓,在2周左右已可完成机化。机化的血栓和血管壁有牢固的粘着,不再有脱落的危险。血栓机化中的新生内皮细胞,被覆血栓内由于血栓干涸产生的裂隙,形成迷路状但可互相沟通的管道,使血栓上下游的血流得以部分地沟通,这种现象称为再通(recanalization)(图3-6)。近年发现,血管腔内的单个核细胞也可自血液内通过血栓的游离面侵入血栓内,而且在血栓内部埋藏着的单个核细胞也可活化,上述的细胞都可游走,被覆于血栓内的裂隙,继而转变成血管内皮细胞,形成新生的血管,所以,血栓机化和再通并不完全依赖于血管壁细胞成分的侵入。
3.钙化长久血栓即不被溶解又不被充分机化时,可发生钙盐沉着。在静脉即形成静脉石(phlebolith)。
四、血栓对机体的影响
血栓形成能对破裂的血管起堵塞破裂口的作用,阻止出血,这是对机体有利的一面,如胃十二指肠慢性溃疡的底部和肺结核性空洞壁,其血管往往在病变侵蚀时已形成血栓,避免了大出血的可能性。然而,在多数情况下,血栓造成的血管管腔阻塞和其他影响,却对机体造成严重的甚至致命的危害。
1.阻塞血管动脉血栓未完全阻塞管腔时,可引起局部器官缺血而萎缩,如完全阻塞或引起必需的供血量不足而又缺乏有效的侧支循环时,可引起局部器官的缺血性坏死。如脑动脉血栓引起脑梗死、心冠状动脉血栓引起心肌梗死,血栓闭塞性脉管炎引起患肢坏疽等。静脉血栓形成后,若未能建立有效的侧支循环,则引起局部淤血、水肿、出血,甚至坏死,如肠系膜静脉血栓可导致出血性梗死。肢体浅表静脉血栓,由于静脉有丰富的侧支循环,通常不引起临床症状。
图3-6 机化的血栓
血管腔内的血栓已为肉芽组织取代有再通现象
2.栓塞在血栓未和血管壁牢固粘着之前,血栓的整体或部分可以脱落,形成栓子,随血流运行,引起栓塞。如栓子内含着细菌,可引起栓塞组织的败血性梗死或栓塞性脓肿。
3.心瓣膜变形心瓣膜血栓机化,可引起瓣膜粘连,造成瓣膜狭窄,如在机化过程中纤维组织增生而后瘢痕收缩,可造成瓣膜关闭不全,见于风湿性心内膜炎和亚急性细菌性心内膜炎。
4.微循环的广泛性微血栓形成,即DIC,可引起全身性广泛出血和休克。
第四节 栓塞
在循环血液中出现的不溶于血液的异常物质,随着血液流动,阻塞血管管腔,这种现象称为栓塞(embolism)。阻塞血管的物质称为栓子(embolus)。最常见的栓子是脱落的血栓,在少见的情况下,脂肪、空气和羊水也可引起栓塞。
图3-7 栓子运行途径与模式图
一、栓子的运行途径
除罕见情况外,栓子一般随血流运行(图3-7)。左心和大循环动脉内的栓子,最终嵌塞于口径与其相当的动脉分支,大循环静脉和右心内的栓子,栓塞肺动脉干或其分支;肠系膜静脉的栓子,引起肝内门静脉分支的栓塞。在有房间隔或室间隔缺损者,心腔内的栓子可由压力高的一侧通过缺损进入另侧心腔,再随动脉栓塞相应的动脉分支。罕见的情况下可发生栓逆向运行,即下腔静脉内的栓子,由于胸、腹腔内压骤然剧增(如咳嗽、呕吐),可逆血流方向栓塞下腔静脉所属的分支。
二、栓塞的类型和对机体的影响
(一)血栓栓塞
由血栓引起的栓塞称为血栓栓塞(thromboembolism),最为常见,占一切栓塞的99%。
1.肺动脉栓塞 血栓栓子约90%以上来自下肢深部静脉,特别是腘静脉、股静脉和髂静脉,偶可来自盆腔静脉,很少来自下肢浅表静脉。较小的栓子栓塞肺动脉小分支,多见于肺下叶,因肺动脉和支气管动脉之间有丰富的吻合支,支气管动脉的血流可以通过吻合支供应该区肺组织,后者可赖以避免梗死;但若栓塞前,肺已有严重淤血,栓塞的局部肺组织尽管有支气管动脉的供血,其血液循环仍不能维持该部肺组织的正常生存,局部遂出现出血性梗死。肺栓塞的影响取决于栓子的大小和数量,栓子体积即使不大,但数量多,广泛地栓塞肺动脉分支;或栓子大,栓塞动脉主干或大分支,患者即发生气促、紫绀、休克,甚至急性呼吸循环衰竭而猝死(图3-8)。巨大的血栓栓子主要来源于下肢静脉,有时来自右心附壁血栓。特别长的栓子可形成骑跨性栓塞阻塞左右肺动脉干。
图3-8 肺动脉血栓栓塞(箭头示)
肺动脉栓塞引起猝死的机制尚不十分明了。一般认为肺动脉主干或大分支栓塞时,一般认为肺动脉主干或大分支栓塞时,肺动脉反射性收缩和血栓栓子内血小板释出的5-羟色胺和凝血恶烷A2引起的支气管和肺泡导管痉挛和肺动脉、心冠状动脉、支气管动脉痉挛,是急性右心衰竭的原因。
2.大循环的动脉栓塞栓子绝大数来自左心的血栓(如亚急性细菌性心内膜炎时心瓣膜赘生物、二尖瓣狭窄时左心房附壁血栓、心肌梗死的附壁血栓),其次为动脉粥样硬化溃疡和主动脉瘤内膜表面的血栓。当有右向左分流的先先性心隔膜缺损时,患者的静脉血栓可以从右心通过该缺损进入左心,造成大循环的动脉的栓塞,称为反常栓塞(paradoxical embolism)。动脉栓塞以下肢、脑、肾、脾为常见,当栓塞的动脉缺乏有效的侧支循环时,则不可避免地引起局部组织的梗死。例如脑底Wills环栓塞,其环状的动脉联系可保证该部任何阻塞皆不导致脑的梗死。但Wills环远端栓塞时,脑梗死则必然发生。肝有肝动脉和门静脉双重血液供应,所以肝动脉分支栓塞很少引起梗死。
(二)脂肪栓塞
含黄骨髓的长骨发生骨折或脂肪组织严重挫伤时,脂肪细胞破裂所释出的脂滴可侵入破裂的血管进入血流;脂肪肝时也可由于上腹部猛烈挤压、撞击,使肝细胞破裂,其所含脂肪也可进入血流。这都可形成脂肪栓塞(fat embolism)而出现症状。但在某些毫无骨折或脂肪组织挫伤的疾患,例如糖尿病时的血脂过高、烧伤、酗洒和慢性胰腺炎等,也常可在尸检时发现无症状的脂肪栓塞,该脂肪栓子的来源是由于血脂过高或精神激烈刺激、过度紧张使呈悬乳状态的血脂不能保持稳定,游离而成的。在任何应激状态均有儿茶酚胺大量分泌,过多动员储备脂肪,增高血脂,形成过多的乳糜微粒,互相融合,乃形成脂肪滴。因此,骨折和创伤引起的脂肪栓塞时,栓子的来源也不完全是组织内的脂肪。
创伤性脂肪栓塞时,栓子随静脉血流到达肺,直径小于20μm的脂滴可通肺泡壁毛细血管经肺动脉和左心,引起全身器官的栓塞,尤其是脑;大于20μm的脂肪栓子则栓塞于肺。脂肪栓塞的组织,栓子量少者可无肉眼变化,只在冰冻切片脂肪下染色下始见小血管腔内有脂滴。较严重者可见肺水肿、出血和肺不张,脑呈水肿和血管周围点状出血。
(三)气体栓塞
多量空气迅速进入血循环或原已溶解于血液内的气体迅速游离,均可形成气体栓塞(air embolism)。前者可见于分娩或流产时,由于子宫强烈收缩,空气被挤入破裂的子宫壁静脉窦;头颈手术、胸壁和肺创伤损伤静脉时,空气也可在吸气时因静脉腔内的负压而被吸入静脉。空气进入右心后,由于心搏动,将空气和心腔内血液搅拌形成大量的泡沫,泡沫状的液体有可压缩性,当心收缩时不被排出而阻塞肺动脉出口,导致猝死。一般迅速进入血循环的空气量在100ml左右时,即可导致心力衰竭,尸检可见右心肺有泡沫状气体存在。进入血循环的空气可引起一些器官的栓塞,气泡激活血小板使之释出5-羟色胺促进血管收缩,而血小板第3因子又促成该血管的血栓形成,从而加重栓塞症状。但如气体量少,可被溶解于血液而不致引起严重后果。
溶解于血液内的气体迅速游离引起的气体栓塞见于减压病(decompression sickness)。沉箱作业的工人,在沉箱内由于气压高,所吸入的空气较多地溶于血液、组织液和脂肪组织内。如从深水中上升到水面常压环境过于迅速,所受外界气压骤然减低,原来溶于血液内的氧、二氧化碳和氮很快游离,形成气泡,氧和二氧化碳可再溶于体液内被吸收,氮则在体液内溶解迟缓,遂在组织和血液内形成小气泡或互相融合成较大的气泡,于是在血管内形成的气体栓塞及其合并的微血栓可引起局部缺血和梗死,组织(主要为肌肉、肌腱、韧带)内的的气泡引起局部症状(关节和肌肉疼痛)。此称沉箱病(caisson disease)。
(四)羊水栓塞
羊水栓塞(amniotic fluid embolism)是分娩过程中一种较罕见的疾患。在分娩过程中,如羊膜破裂,尤其又有胎儿头阻塞阴道口时,子宫收缩可将羊水压入破裂的子宫壁静脉窦内,羊水成分可由子宫静脉进入肺循环,在肺动脉分支及毛细血管内引起羊水栓塞。少量羊水可通过肺毛细血管进入大循环引起多数器官小血管的栓塞。镜下,羊水栓塞的证据是在小动脉和毛细血管内发现羊水成分:角化上皮,胎毛,胎脂,胎粪和粘液(图3-9)。本病发病急骤,产妇出现紫绀、呼吸困难和休克,绝大多数导致死亡。羊水成分栓塞肺血管所致的肺循环机械性阻塞,实不足以解释上述症状,因此过敏性休克、DIC、羊水液体内所含的血管活性物质进入血液引起血管反应可能是致死的原因。羊水具有凝血致活酶作用,可引起DIC,一些羊水栓塞病例,肺微血管内有纤维素性血栓存在。
图3-9 肺羊水栓塞
小血管内有角化上皮
(五)其他栓塞
肿瘤细胞栓塞,可引起肿瘤在局部形成转移瘤(图3-10)。寄生虫、虫卵和其他异物偶可进入血循环引起栓塞。
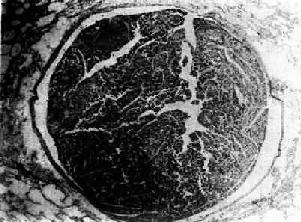
图3-10 肺小动脉腔内肝癌细胞栓子
第五节 梗死
器官或组织的血液供应减少或停止称为缺血(ischemia),凡动脉腔狭窄或闭塞而没有充分的侧支循环或侧支循环不能及时建立时,局部组织即发生缺血。由于缺氧、营养物质输入不足或停顿和代谢中间产物聚积,组织即可出现病理改变,其程度决定于缺血的程度和受累组织对缺氧的耐受性。一般而言,缺血程度较轻和持久时,实质细胞发生萎缩和变性,严重而迅速发生的缺血则引起组织坏死。缺血与坏死有密切的因果关系,坏死的原因很多,其中由于血管阻塞引起的组织坏死灶称为梗死(infarct)。
一、梗死的原因
任何引起血管管腔阻塞,导致局部组织缺血的原因均可引起梗死。
1.血栓形成是梗死的最常见原因,如心冠状动脉和脑动脉的粥样硬化合并血栓形成,可分别引起心肌梗死和脑梗死,趾、指的血栓闭塞性脉管炎引起趾、指梗死(坏疽)等。
2.动脉栓塞也是梗死的常见原因,在肾、脾和肺的梗死中,由栓塞引起者远比动脉血栓形成引起者多见。
3.血管受压闭塞动脉受肿瘤或其他机械性压迫而致管腔闭塞时可引起局部组织梗死,肠套叠、肠扭转和嵌顿性疝时肠系膜静脉受压,血液回流受阻,同时肠系膜动脉亦因受压而致输入血量不同程度地减少,局部组织血液循环停顿,亦可引起肠梗死。
4.动脉痉挛近年来注意到,在已有心冠状动脉硬化时,如发生动脉痉挛,亦可能引起心肌梗死。
二、梗死的病变
梗死是局限性组织坏死,梗死的形状决定于该器官的血管分布。多数器官的血管呈锥形分支,如脾、肾、肺等,故其梗死也呈锥形,切面呈扇面形,其尖端位于血管阻塞处,底部则为该器官的表面(图3-11)。心冠状动脉分支不规则,故心肌梗死形状亦不规则或呈地图状。梗死灶的质地决定于其坏死的类型。梗死灶为凝固性坏死者(肾、脾、心肌),新鲜时由于组织崩解,局部胶体渗透压升高而吸收水分,使局部肿胀,略向表面隆起,切面可略凸出。陈旧性梗死则较干燥,质硬,表面下陷。脑梗死为液化性坏死,新鲜时质地软、疏松,日久液化成囊。梗死灶的颜色取决于病灶内的含血量,含血量少者,颜色灰白,称为贫血性梗死(anemic infarct);含血量多者,颜色暗红,称为出血性梗死(hemorrhagic infarct)。
1.贫血性梗死发生于组织结构比较致密,侧支循环不充分的器官,如肾、脾、心肌(图3-12)。当其

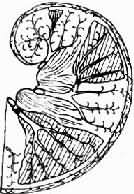
图3-11 肾动脉分支栓塞及肾贫血性梗死(模式图)
动脉分支阻塞时,局部组织立即陷于缺氧而使其所属微血管通透性增高,病灶边缘侧支血管内的血液可通过该通透性增高的微血管壁逸出于血管外,即出血。在肾、脾、心肌等器官,由于组织致密,故出血量不多,出血的红细胞崩解后,血红蛋白溶于组织液被吸收,故梗死灶呈灰白色。在梗死的早期,梗死的周围有明显的充血和出血,形成暗红色出血带,数日后该出血带内的红细胞已被巨噬细胞吞噬后转变为含铁血黄素,出血带遂变为褐黄色。镜下,早期的梗死灶内尚见核固缩、核碎裂和核溶解等变化,细胞浆则均匀一致,组织结构仅见其粗略轮廓。病灶内可见橙色血晶。晚期,病灶表面下陷,质地坚实,原已呈褐黄色的出血带亦消失。镜下,病灶呈均质性结构,边缘有肉芽组织和瘢痕形成。梗死灶小者,可完全被肉芽组织和瘢痕组织所取代。
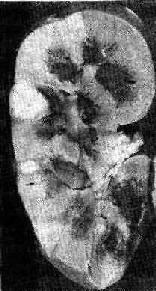
图3-12 肾贫血性梗死
肾皮质可见二个梗死灶,略呈楔形,底部靠肾表面,尖指向肾门
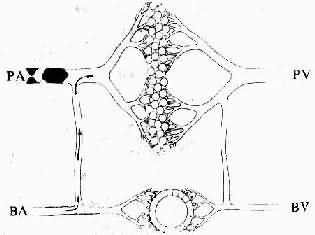
图3-13 肺动脉栓塞时的血流变化示意图
PA:肺动脉;PV:肺静脉;BA:支气管动脉;BV;支气管静脉(仿Eder)
2.出血性梗死 出血性梗死发生于下述条件下:
(1)严重淤血:当器官原有严重淤血时,血管阻塞引起的梗死为出血性而非贫血性。如卵巢肿瘤在卵巢蒂扭转时,由于静脉回流受阻,动脉供血随之停止,卵巢瘤组织随即坏死,同时血液由淤血的毛细血管内漏出,形成出血性梗死。肺梗死多发生于患者已有左心功能代偿不全的先决条件下,此时的肺淤血是梗死灶内发生出血的原因。
(2)组织疏松:肠和肺的组织较疏松,梗死初起时在组织间隙内可容多量出血,当组织坏死而膨胀时,也不能把漏出的血液挤出梗死灶外,因而梗死灶为出血性。但如肺先因肺炎而实变,则所发生的肺梗死一般为贫血性而非出血性。
需指出的是,肺梗死发生的先决条件为事先需有肺淤血的存在。这是因为肺有肺动脉和支气管动脉双重血液供应,两者之间有丰富的吻合支,有肺循环正常的条件下,肺动脉分支栓塞不会引起梗死,因为支气管动脉可借助吻合支供血于该区肺组织(图3-13);但如肺原先已有淤血,致肺静脉压增高,当肺动脉分支栓塞时,单纯以支气管动脉的压力不足以克服局部范围内的肺静脉阻力,局部肺组织乃发生梗死。这便是肺梗死常见于二尖瓣疾患而且是出血性的原因。
肺的出血性梗死为底靠肺膜、尖指向肺门的锥形病灶(图3-14),暗红色,出血性梗死组织之镜下结构为组织坏死伴有弥漫性出血。

图3-14 肺出血性梗死
肺组织内见一典型的楔形梗死区,梗死区内肺组织出血、坏死(图内呈黑色)
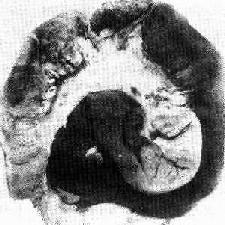
图3-15 小肠出血性梗死
梗死的肠壁呈暗红色
出血性梗死亦常发生于肠。肠套叠、肠扭转、嵌顿性疝均可引起局部肠段出血性梗死,肉眼观该肠段呈暗红色(图3-15)。
梗死又可按有无细菌感染而分为败血性梗死(septic infarct)和单纯性、无感染性梗死。前者的栓子含有细菌,因而梗死灶内有细菌感染。急性细菌性心内膜炎时,由心瓣膜脱落的含细菌栓子造成栓塞时可引起栓塞性脓肿。
三、梗死的影响和结局
梗死对机体的影响决定于发生梗死的器官和梗死灶的大小和部位。肾有较大的代偿功能,肾梗死通常只引腰痛和血尿,但不影响肾功能。四肢的梗死即坏疽,可引起毒血症,必要时须截肢。肺梗死有胸膜刺激征和咯血。心肌梗死可影响心功能,严重者可致心功能不全。脑梗死视不同定位而有不同症状,梗死灶大者可致死。
梗死灶形成时,病灶周围的血管扩张充血,并有白细胞和巨噬细胞渗出,继而形成肉芽组织。在梗死发生24~48小时后,肉芽组织已开始从梗死灶周围长入病灶内,小的病灶可被肉芽组织所取代,日后变为瘢痕。大的梗死灶不能完全被机化时,则由肉芽组织和日后转变成的瘢痕组织加以包裹,病灶内部则可钙化。脑梗死则液化成囊腔,周围由增生的胶质瘢痕包裹。
第六节 水肿
组织间隙或体腔内过量的体液潴留称为水肿(edema),然而通常所称的水肿乃指组织间隙内的体液增多,体腔内体液增多则称积水(hydrops)。水肿可表现为局部性或全身性,全身性水肿(anasarca)时往往同时有浆膜腔积水,如腹水(ascites)、胸腔积水(hydrothorax)和心包腔积水(hydropericardium)。
一、水肿的原因和机制
在生理状态下,血液的流体静力压及组织液的胶体渗透压和血浆的胶体渗透压及组织液的流体静力压是两组互相拮抗的压力,这两组压力保持动态平衡。组织液胶体渗透压、血浆胶体渗透压和组织液流体静力压皆稳定,分别为1.3kPa(10mmHg)、3.4kPa(25mmHg)和0.66kPa(5mmHg),而血液的流体静力压则在毛细血管动脉端和毛细血管静脉端有明显差别,前者为4.3kPa(32mmHg),后者为1.6kPa(12mmHg)。在毛细血管动脉端,血管内流体静压与组织液胶体渗透压之和大于血浆胶体渗透压与组织液流体静力压之和,所以液体从毛细血管内移向血管外;在毛细血管静脉端则相反,血浆胶体渗透压与组织液流体静力压之和大于血管内流体静力压与组织液胶体渗透压之和,液体从组织间隙移入毛细血管内。少部分组织液可通过淋巴管回纳入血液,组织液的形成和吸收处于动态平衡。水肿形成的机制,即上述平衡失调,体液自血管内逸出到组织间隙过多和(或)体液自组织间隙回纳入血液过少。概括而言,引起平衡失调的原因不外①血浆胶体渗透压降低,②毛细血管内流体静力压升高,③毛细血管壁通透性增高,④淋巴液回流受阻。
1.血浆胶体渗透压降低见于蛋白质吸收不良或营养不良及伴有大量蛋白尿的肾脏疾患等。当血浆白蛋白量降到2.5±0.5g%或总蛋白量降到5±0.5g%时,就可出现水肿,为全身性。
2.毛细血管内流体静力压升高见于各种原因引起的静脉阻塞或静脉回流障碍。局部静脉回流受阻引起相应部位的组织水肿或积水,如肝硬变引起胃肠壁水肿的和腹水,心力衰竭时的腔静脉回流障碍则引起全身性水肿。
3.毛细血管壁通透性增高血管活性物质(组胺、激肽)、细菌毒素、缺氧等可增加毛细血管壁的通透性而引起水肿。炎性病灶的水肿即主要由于毛细血管壁的通透性增高,血管神经性水肿和变态反应引起的水肿亦属此一机制。此类水肿通常发生于血管壁受损的局部。
4.淋巴回流受阻乳腺癌根治术后,由于腋窝淋巴结切除后的局部淋巴液循环破坏,可发生患侧上肢水肿;丝虫病时下肢和阴囊由于淋巴管被虫体阻塞,常发生下肢和阴囊水肿。此外淋巴管广泛性的癌细胞栓塞可引起局部水肿。
在心力衰竭、肝硬变、肾病综合征的水肿形成中,肾素-血管紧张素-醛固酮系统起了辅助作用。心力衰竭时的心搏出量减少,肾灌注血量不足,刺激肾近球装置,使肾素分泌增多,后者使血管紧张素原变为有活性的血管紧张素Ⅰ,再经转换酶的作用将血管紧张素Ⅰ变为血管紧张素Ⅱ,后者作用于肾上腺皮质球状带细胞,使之分泌醛固酮,从而促进肾远曲小管的钠重吸收,招致钠潴留,引起血液晶体渗透压增高,后者刺激血管壁渗透压感受器,使垂体后叶分泌抗利尿激素,从而加强肾远曲小管的水重吸收。水的潴留助长了心源性水肿的形成。肝硬变时的水肿和腹水,也有醛固酮的作用参与,这是由于肝细胞对醛固酮的灭活作用减退,同时,在腹水形成之后,由于循环血量减少,又引起醛固酮分泌增多。肾病综合征因白蛋白大量流失,血浆蛋白量低落,发生水肿,体液自血管内向血管外逸出,循环血量下降,又激发肾素-血管紧张素-醛固酮系统的活性。
二、水肿的病变
发生水肿的组织,体积增大,颜色苍白,镜下可见水肿液积于细胞和纤维结缔组织之间或腔隙内。由于水肿液含血浆蛋白,故HE染成粉红色。肺水肿时,肺泡腔内充满水肿液(图3-16)。切开肺时可有泡沫状液体自切面溢出。脑水肿时脑回变扁平,脑沟变浅。镜下,脑灰质和白质疏松,血管周围间隙加宽。严重时脑组织在高倍镜下呈网化状态。严重脑水肿时可形成脑疝。
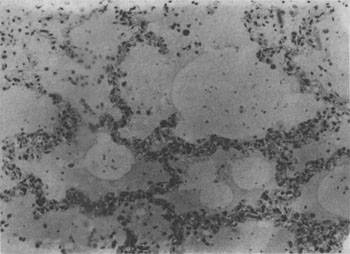
图3-16 肺水肿
肺泡腔内有水肿液及漏出的红细胞,肺泡壁毛细血管扩张充血
第四章 免疫病理
免疫反应是机体在进化过程中所获得的“识别自身、排斥异己”的一种重要生理功能,在正常情况下,免疫系统通过细胞和(或)体液免疫机制以抵抗外界入侵的病原生物;维持自身生理平衡;以及消除突变细胞,起到保持机体的作用。但免疫反应异常,无论是反应过高(变态反应)或过低(免疫缺陷)均能引起组织损害,导致疾病,迄今公认为的免疫性疾病已不数十种,本章于介绍免疫损伤基本病变的基础上,重点途述移植排斥反应以及常见的几种自身免疫性疾病和免疫缺陷病。
第一节 组织损伤的免疫机制
由内源性或外源性抗原所致的细胞或体液介导的免疫应答导致的组织损伤称免疫损伤(immune injury)通常称之为变态反应(allergic reaction)或超敏反应( hypersensitivity reaction)。引起免疫性损伤的抗原可以是内源的或外源的,同种的或自体的,其中来自外环境的外源性抗原所致的过敏反应有些是可以预防的,如接触毒葛所致的接触性皮炎,接触花粉所致的枯草热等,均可通过避免接触抗原加以预防。部分同种抗原所致的过敏反应如输血反应,通过受、供血液的交叉配型亦可以避免。
变态反应按免疫机制的不同可分为四类,即Ⅰ、Ⅱ、Ⅲ、Ⅳ型。
一、Ⅰ型变态反应
Ⅰ型变态反应又称过敏反应(anaphylaxis),因反应迅速,故又有速发型超敏反应(immediate hypersensitivity)之称。本型变态反应是通过抗原(致敏原)进入机体后与附着在肥大细胞和嗜碱性粒细胞上的IgE分子结合,并触发该细胞释放生物活性物质,引起平滑肌收缩、血管通透性增加、浆液分泌增加等临床表现和病理变化。
致敏原的种类繁多,常见的有①异种蛋白质:如异种动物血清、蜂毒、昆虫毒液、疫苗、寄生虫、食物、花粉、胰岛素等;②药物:如各种抗生素、有机碘、汞剂等。
【发病机制】
Ⅰ型变态反应在人类乃由IgE抗体所介导。致敏原刺激扁桃体、肠的集合淋巴结或呼吸道粘膜中的淋巴细胞、巨噬细胞、在TH细胞的协同作用下,产生IgE(在一般情况下,这一过程受TS细胞的抑制)。IgE的Fc片段与肥大细胞嗜碱性粒细胞的Fc受体相结合,造成了致敏状态。当机体再次接触相同的致敏原时,它们与附着于肥大细胞上之IgE相结合。多价抗原与二个以上领近的Ige 分子发生交联,激发了二个平行但又独立的过程,其一是肥大细胞的脱颗粒和颗粒中介质(原发性介质)的释放;另一是细胞膜中原位介质合成和释放(图4-1)。
1.原发性介质存在于肥大细胞的颗粒中,通过脱颗粒而释放,主要包括①组胺,可引起强烈的支气管平滑肌收缩,血管扩张、通透性增加,粘液分泌增加;②趋化因子(chemotactic factor, CF),其中嗜酸性粒细胞趋化因子(ECF-A)和中性粒细胞趋化因子(NCF)分别引起嗜酸性粒细胞和中性粒细胞浸润;③中性蛋白酶可裂解补体及激肽原而产生其他炎症介质。
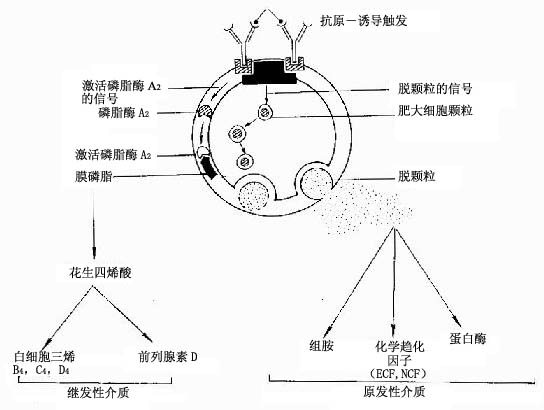
图4-1 Ⅰ型变态反应中肥大细胞释放介质的机制
2.继发性介质由激活的肥大细胞所产生,主要通过磷脂酶A2的激活,作用於膜磷脂而产生花生四烯酸,进而通过5-脂氧化酶和环氧化酶途径分别产生白细胞三烯(leucotrienes,LT)和前列腺素(prostaglandins,PG):①白细胞三烯是最强烈的血管活性和致痉物质,其效应较组胺高数千倍,而LTB4对中性、嗜酸性粒细胞及单核细胞有很强的趋化性;②前列腺素D2(PGD2)多产生于人肺肥大细胞,可引起强烈的支气管痉挛和粘液分泌增多;③血小板激活因子(platelet activating factor, PAF)可引起血小板聚集和组胺释放,该因子的产生也是磷脂A2激活所致,但并非花生四烯酸的代谢产物。此外肥大细胞尚可分泌多种细胞因子,其中如TNF-α对促进炎细胞浸润亦起重要作用。
Ⅰ型变态反应过程无补体参与,在一般情况下不破坏细胞,其致病作用主要是通过上述各生物活性物质引起。本型反应有局部性和全身性两类;:局部性反应常表现为局部组织水肿、嗜酸性粒细胞浸润、粘液分泌增加或支气管平滑肌痉挛等变化,如皮肤荨麻疹(食物过敏),过敏性鼻炎(枯草热)及哮喘等。而全身性过敏反应如抗血清、药物(如青霉素)的过敏性休克,可造成迅速死亡。死亡病例尸检时,可见喉头水肿,两肺出血水肿,有时伴急性肺气肿及右心扩张,血不凝固,其余内脏除淤血外,通常无特征性形态变化。
二、Ⅱ型变态反应
Ⅱ型变态反应又名细胞毒性抗体反应,是由抗体与靶细胞表面的抗原相结合而介导。抗原可以是细胞膜自身成分,也可以是吸附在细胞表面的外源性抗原或半抗原,可通过不同的机制而引起细胞损害。
1.补体介导的细胞毒反应(complement mediated cytotoxicity,CMC) 特异性抗体(IgM或IgG)与细胞表面的抗原相结合,固定并激活补体,直接引起细胞膜的损害与溶解,或通过抗体的Fc片段及C3b对巨噬细胞相应受体的亲和结合,由巨噬细胞所介导。此反应常累及血细胞(红细胞、白细胞、血小板)和细胞外组织如肾小球基底膜,引起细胞和组织损害。
临床上此类Ⅱ型变态反应常见于下列情况:①血型不符的输血反应,这是由于供者红细胞抗原与受者血清中的相应抗体相结合而导致红细胞的溶解;②新生儿溶血性贫血(胎儿有核红细胞增多症)是由于母体(Rh阴性)和胎儿(Rh阳性)抗原性差异所致,母体产生的抗Rh抗体(IgG)通过胎盘引起胎儿红细胞破坏,导致溶血;③自身免疫性溶血性贫血、粒细胞减少症、血小板减少性紫癜等疾病,它们是由于不明原因引起自身血细胞抗体形成而导致相应血细胞的破坏;④某些药物反应,药物作为半抗原与血细胞膜结合形成抗原,激发抗体形成,后者针对血细胞-药物复合物(抗原)而引起血细胞的破坏。
2.依赖抗体介导的细胞毒反应(antibody dependent cellular cytotoxity,ADCC) 在本反应中,靶细胞为低浓度的IgG抗体所包绕,IgG的Fc片段可与一些具有Fc受体的细胞(K细胞、中性粒细胞、嗜酸性粒细胞、单核细胞)相接触而引起靶细胞的溶解(图4-2),后者需要消耗能量但不涉及吞噬反应或补体的固定。ADCC反应主要与寄生虫或肿瘤细胞的消灭以及移植排斥有关。
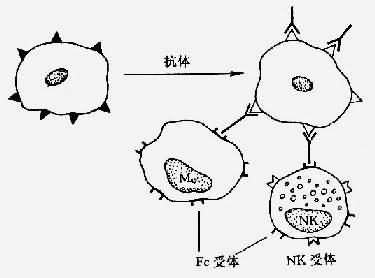
图4-2 抗体依赖细胞介导细胞毒作用(ADCC)示意图
3.抗体介导的细胞功能异常患者体内存在抗某种受体的自身抗体,抗体与靶细胞表面的特异性受体结合从而导致靶细胞的功能异常。由于不结合补体,因而不破坏靶细胞亦无炎症反应。例如重症肌无力(myasthenia gravis),是由于患者体内存在抗乙酰胆碱受体的自身抗体,此抗体可与骨骼肌运动终板突触后膜的乙酰胆碱受体结合,削弱神经肌冲动的传导而导致肌肉无力。
三、Ⅲ型变态反应
Ⅲ型变态反应又名免疫复合物介导的超敏反应(immune complex mediated hypersensitivity)。免疫复合物是抗原和抗体相结合的产物,在生理情况下它能及时被吞噬系统所清除。如免疫复合物是沉积于血管壁进而引起血管炎症,则导致疾病。引起人体免疫复合物疾病的抗原种类繁多,有微生物(细菌、病毒等)、寄生虫、异体蛋白(食物、血清等)、药物(青霉素、普鲁卡因酰胺等)、自身抗原(变性IgG、核酸等)、肿瘤抗原(肿瘤相关抗原、癌胚抗原等)及其他原因不明性抗原。至于抗体则限于能被补体固定的IgG和IgM,而非IgA、IgD或IgE。
免疫复合物沉积引起组织损伤的主要环节是固定并激活补体,产生生物活性介质,而导致组织损伤及炎症反应,包括:
1.补体激活补体激活所产生的生物学效应有:①通过释放C3b促进吞噬作用;②提供趋化因子(C5b、C567),诱导中性粒细胞和单核细胞游走;③释放过敏毒素(C3a,C5a),增加血管通透性和引起平滑肌收缩;④攻击细胞膜造成细胞膜损伤甚至溶解(C5b~9复合体)。
2.血小板聚集和Ⅻ因子激活免疫复合物可引起血小板聚集和Ⅻ因子(Hageman factor)激活,两者均可促进炎症过程和微血栓形成,从而导致缺血和坏死。
3.炎症介质释放白细胞吞噬抗原抗体复合物后可释放多种炎症介质,包括前列腺素、扩张血管的肽类物质、阳性趋化物质以及多种溶解体酶,其中的蛋白酶能消化基底膜、胶原、弹力纤及软骨。此外,激活的中性粒细胞产生的氧自由基也可引起组织损害。
Ⅲ型变态反应因复合物沉积部位的不同,导致的免疫复合物病有局限性与全身性两类。
局限性免疫复合物沉积引起的变态反应又称Arthus 反应,是急性免疫复合物性血管炎所致的局部组织坏死,常发生在皮肤。实验性Arthus反应可采用具有循环抗体的免疫动物以相应的抗原作皮下注射来引起,当注射在皮下的抗原弥散到局部血管时,即可与血液中的抗体相结合而形成免疫复合物沉积于血管壁,激发炎症反应,表现为局部水肿、出血和坏死,血管壁纤维素样坏死明显,常伴有血栓形成,局部的缺血更加重了组织的损害。
全身性免疫复合物病又称血清病(serum sickness),乃因抗原抗体在循环中形成可溶性复合物在组织中沉积而致病。其中,一次大量免疫复合物形成并在多器官沉积,引起急性血清病;而反复持续沉积则引起慢性血清病。血清病常累及的部位是肾、心血管、关节滑膜、皮肤等血管丰富的组织。
急性血清病的临床表现根据免疫复合物的量和形成的速度而异。当大量异体蛋白注入含有大量相应抗体的血液时,迅速形成高浓度的免疫复合物,可导致虚脱。这是由于补体被激活,中性粒细胞释放溶酶体酶,血小板激肽的释放,以及凝血与纤溶系统被激活所致。临床表现与过敏性休克相似。经典的血清病是在首次注射异体蛋白7~10天(产生抗体)后出现,临床上表现为短暂的发热、皮肤荨麻疹、周围关节肿胀、淋巴结肿大和蛋白尿等,血清补体含量明显降低。免疫复合物沉积所致的血管病表现为管壁纤维素样坏死伴大量中性粒细胞浸润,免疫荧光或免疫电镜检查可证实管壁有抗体和补体存在。
慢性血清病是在持久抗原血症,免疫复合物形成和沉积所致。慢性免疫复合物病最常累及肾,引起膜性肾小球肾炎。
四、Ⅳ型变态反应
Ⅳ型变态反应又名迟发型超敏反应,是特异性致敏T细胞所介导,其中包括经典的迟发型超敏反应和细胞介导的细胞毒性反应,两者均系致敏T细胞接触特异性抗原而引起,分别受到MHC-Ⅱ类和MHC-Ⅰ类抗原的限止。在迟发型超敏反应中,有其他细胞的参与,其中巨噬细胞为主要的效应细胞。在细胞介导的细胞毒性反应中,致敏T细胞本身具有效应功能,可直接攻击靶细胞。Ⅳ型变态反应是各种细胞内感染,特别是结核杆菌、病毒、真菌和寄生虫感染所致的免疫反应。其他如化学物质所引起的接触性皮炎及移植排斥也属本型反应。
1.迟发型超敏反应迟发型超敏反应最经典的例子是结核菌素反应。结核菌素是结核菌的蛋白脂多糖成分。对先前已致敏的个体,皮内注射结核菌素,8~12小时后局部出现硬结。一般在1~3天达到高峰。镜下可见表皮和真皮浅部有多量单核细胞的聚集和多少不等的中性粒细胞浸润,血管通透性明显增高,间质中有较多纤维蛋白的沉积。
本反应的发生主要由CD4+T细胞介导,当机体再次接触特异性抗原时,致敏CD4+细胞受刺激,激活分裂并释放各种生物活性物质,名为淋巴因子(lymphokines),淋巴因子和其他细胞因子(cytokines)一样,通过三种方式产生效应,以促成迟发型超敏反应的发生,即①自分泌(autocrine)机制,CD4+细胞分泌的白介素2(IL-2)作用于CD4+细胞表面的IL-2受体而使CD4+细胞本身进一步激活、增生并分泌;②旁分泌(paracrine)机制,CD4+细胞分泌的干扰素(IFN-γ)等作用于邻近的巨噬细胞,将其激活、聚集并分泌单核因子如IL-1、PDGF等,导致肉芽肿性炎症的形成和发展;③内分泌(endocrine)机制,使远处或系统细胞产生效应,例如CD4+细胞等分泌的肿瘤坏死因子(TNF-α)和淋巴毒因子(lymphotoxin)可作用于血管内皮细胞,前者增加其前列环素的分泌,使血管扩张,后者使之表达淋巴细胞粘附分子(ELAM-1)有利于淋巴、单核细胞粘附并游出。
2.细胞毒反应由CD8+T细胞所介导,此种淋巴细胞又名细胞毒性T细胞(CTL),其溶细胞作用是通过CTL表面的特异性受体对靶细胞表面抗原的识别、结合而受启动,通过细胞与细胞的直接接触,最终导致靶细胞膜的溶解和细胞的坏死。
因此总的说来,Ⅳ型变态反应病变的特点是以单个核细胞浸润为主的炎症和组织坏死。如果局部有些难以降解的抗原刺激持续存在,则在单个核细胞浸润的基础上,数周后可出现类上皮细胞结节,形成典型的肉芽肿。胞内寄生菌(如结核杆菌、麻风杆菌),某些真菌(如荚膜组织胞浆菌、新型隐球菌),某些寄生虫(如血吸虫卵的可溶性抗原)感染以及病素感染,器官移植排斥反应,肿瘤免疫等常出现明显的Ⅳ型变态反应。
需要指出的是免疫反应可通过上述四大类机制引起病变,但以一个疾病而言,由于抗原的特性,机体的反应情况以及病程发展的不同阶段,可以同时或先后出现不同类型的变态反应,将在以下几种疾病中进一步说明。
第二节 移植排斥反应
在同种异体组织、器官移植时,受者的免疫系统常对移植物产生排异反应(transplant rejection),这是一个十分复杂的免疫学现象,涉及细胞和抗体介导的多种免疫损伤机制,都是针对移植物中的人类主要组织兼容抗原HLA(human leucocyte antigen) 的反应,供者与受者HLA的差异程度决定了排异反应的轻或重。除单卵双生外,二个个体具有完全相同的HLA系统的组织配型几乎是不存在的,但选择供者与受者配型尽可能地接近,是异体组织器官移植成功的关键。
HLA系统是目前人类已知的最复杂的基因群—HLA复合体,或称MHC基因的编码产物。HLA复合体位于第六对染色体的短臂,是免疫遗传的调控中心。每个基因位点存在多种等位基因,导致HLA系统的高度多态性。HLA复合体的基因位点,从功能角度可分为三类。第一类:包含基因位点HLA-A·B·C,所有真核细胞均具有Ⅰ类基因位点。第二类即HLA-D(DR)位点,主要见于抗原递呈细胞、B细胞及T辅助细胞,此外,血管内皮细胞、纤维母细胞及肾小管上皮细胞等如果受到淋巴因子IFN-γ的诱导,亦可表达Ⅱ类抗原。第三类则属于编码补体C2、C4和B因子的基因位点。HLA分子的主要生理作用是将抗原递呈给T细胞,因而无论对诱导体液及细胞免疫反应均起重要作用。已知调控免疫反应的Ir(immune response)基因亦位于HLA-D区,所以Ⅱ类抗原的基因亦具有Ir基因的功能,调控免疫反应的幅度。因而可以认为与HLA直接相关的免疫病理现象是:移植排斥反应、对疾病的反应过度或反应不足。
一、排斥反应的机制
移植排斥反应过程很复杂,既有细胞介导的又有的抗体介导的免疫反应参与作用。
1.T细胞介导的排斥反应在人体和实验性组织、器官移植中证实,T细胞介导的迟发性超敏反应与细胞毒作用对移植物的排斥起着重要的作用。移植物中供体的淋巴细胞(过路淋巴细胞)、树突细胞等具有丰富的HLA-Ⅰ、Ⅱ抗原,是主要的致敏原,它们一旦被受体的淋巴细胞识别,即可引起以下系列变化(图4-3)。
(1)CD8+细胞毒性T细胞(CTL)前细胞:前细胞具有HLA-I受体,与HLA-i 抗原结合后可引起分化,成为成熟的CTL,溶解移植组织。
(2)CD4+T辅助细胞(TH):TH细胞能识别HLA-Ⅱ抗原并与之发生作用引起移植物中抗原递呈细胞释放白细胞介素I(IL-I),后者可促进TH细胞增生和释放IL-2,而IL-2可进而促进TH细胞增生并为CTL细胞的分化提供辅助信号。除了IL-2之外,TH细胞还能产生IL-4、IL-5、促进B细胞分化并产生抗移植物的抗体,参与移植排斥。此外与迟发变态反应相拌随的血管损害、组织缺血及巨噬细胞介导的破坏作用,也是移植物毁损的重要机制。
2.抗体介导的排斥反应T细胞在移植排斥反应中无疑起着主要作用,但抗体也能介导排斥反应,其形式有二:①过敏排斥反应,发生在移植前循环中已有HLA抗体存在的受者。该抗体来自过支曾多次妊娠、接受输血、人工透析或感染过某些其表面抗原与供者HLA有交叉反应的细菌或病毒。在这种情况下,器官移植后立即可发生排斥反应(超急性排斥),此乃由于循环抗体(抗HLA)固定于移植物的血管内皮(表达HLA)发生Ⅱ型变态反应,引起血管内皮受损,导致血管壁的炎症、血栓形成和组织坏死;②在原先并无致敏的个体中,随着T细胞介导的排斥反应的形成,可同时有抗HLA抗体形成,此抗体在移植后接受免疫抑制治疗的患者中对激发晚期急性排斥反应颇为重要。免疫抑制药虽能一定程度上抑制T细胞反应,但抗体仍在继续形成,并能过补体介导的细胞毒(CMC)、依赖抗体介导的细胞毒(ADCC)及抗原抗体免疫复合物形成等方式,引起移植物损害。
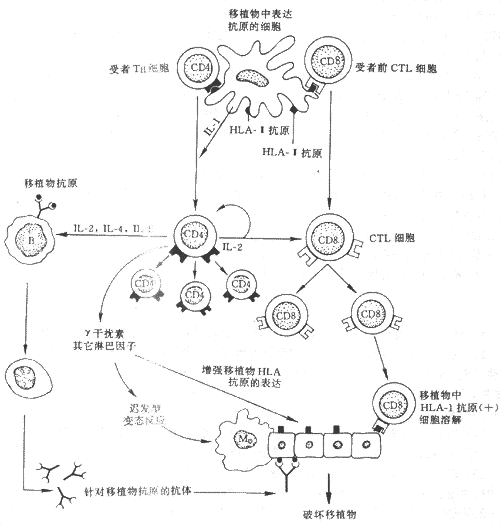
图4-3 组织不相容性移植物破坏过程示意图
二、排斥反应的病变
移植排斥反应按形态变化及发病机制的不同有超急性排斥反应、急性排斥反应和慢性排斥反应三类。兹以肾移植中各类排斥反应的病理变化为例加以说明。类似的变化亦可见于其他组织、器官的移植。
1.超急性排斥反应是受体对移植物的一种迅速而剧烈的反应,一般于移植后数分钟至24小时内出现.本型反应的发生与受体血循环中已先有供体特异性HLA抗体存在,或受体、供体ABO血型不符有关.这主要是由于循环抗体与移植物细胞表面HLA抗原相结合并激活补本系统,释放出多种生物活性物质,从而引起局部炎症、血管内皮细胞损害、血栓形成和组织损伤。本质上属Ⅱ型变态反应,但广泛分布的急性小动脉炎、血栓形成和因而引起的组织缺血性坏死,在形态上类似于Arthus反应。
移植肾肉眼观表现为色泽迅速由粉红色转变为暗红色,伴出血或梗死,出现花班状外观。体积明显肿大,质地柔软,无泌尿功能。镜下表现为广泛的急性小动脉炎伴血栓形成及缺血性坏死。受累的动脉壁有纤维素样坏死和中性粒细胞浸润,并有IgG、IgM、补体沉积;管腔中有纤维蛋白和细胞碎屑阻塞。肾小球肿大,肾小管上皮细胞发生缺血性坏死,间质水肿并有中性粒细胞浸润,有时还可有淋巴细胞和巨噬细胞浸润。
2.急性排斥反应较常见,在未经治疗者此反应可发生在移植后数天之内;而经过免疫抑制治疗者,可在数月或数年后突然发生。此种排斥反应可以细胞免疫为主,也可以体液免疫为主,有时两者可同时参与作用。
(1)细胞型排斥反应:常发生在移植后几个月,临床上表现为骤然发生的移植肾功能衰竭。镜下可见肾间质明显水肿伴有大量细胞浸润,以单核细胞和淋巴细胞为主,并夹杂一些具有嗜酸性胞浆和水泡状胞核的转化淋巴细胞和浆细胞。免疫组化染色证实有大量CD4+、CD8+细胞存在。肾小球及肾小管周围毛细血管中有大量单核细胞,间质中浸润的淋巴细胞可侵袭肾小管壁,引起局部肾小管坏死(图4-4)。
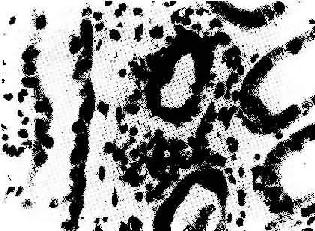
图4-4 肾移植急性排斥反应
肾间质水肿,肾小管上皮细胞变性,间质及毛细血管内有大量单核细胞
(2)血管型排斥反应:主要为抗体介导的排斥反应(CMC,ADCC和免疫复合物形成),往往在稍后出现,以突出的血管病变为特征。表现为肾细、小动脉的坏死性血管炎,可呈弥漫或局灶性分布。免疫荧光证实有免疫球蛋白、补体及纤维蛋白沉积,肾小球毛细血管袢亦可受累。纤维蛋白样坏死的血管壁内常有淋巴细胞、泡沫细胞及中性粒细胞浸润,腔内可有血小板凝集、血栓形成(图4-5)。后期的血管内膜纤维化,管腔狭窄。间质内常有不同程度淋巴细胞、巨噬细胞及浆细胞浸润。肉眼观,肾常明显肿大,呈暗红色并有出血点,有时可出现黄褐色的梗死灶,可伴有肾盂及肾盏出血(图4-6).临床上,移植肾出现功能减退,大剂量免疫抑制剂疗效不佳。
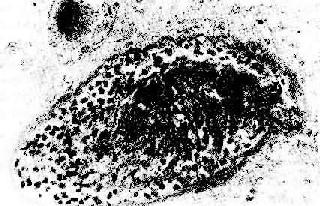
图4-5 肾移植急性排斥反应
小叶间动脉呈纤维素样坏死,并有大单核细胞及中性白细胞浸润和血栓形成
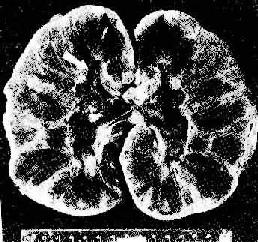
图4-6 肾移植急性排斥反应
肾肿胀,明显出血,肾盏及肾盂中充满血块
3.慢性排斥反应慢性排斥是反复急性排斥的积累,其突出的病变是血管内膜纤维化,常累及小叶间弓形动脉。动脉内膜纤维化引起管腔严重狭窄(图4-7),导致肾缺血,表现为肾小球毛细管袢萎缩、纤维化、玻璃样变,肾小球萎缩,间质除纤维化外尚有中等量单核、淋巴细胞浸润。肉眼观,肾体积明显缩小,并有多少不等的瘢痕(“小瘢痕肾”)。包膜明显增厚并有粘连。患者肾功能呈进行性减退,其程度与间质纤维化和肾小球、肾小管萎缩的程度成正比。
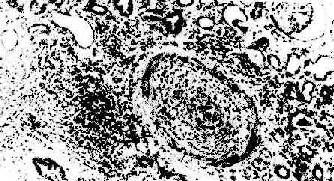
图4-7 肾移植慢性排斥反应
小叶间动脉管壁纤维化,管腔闭塞,内膜有中等量单核细胞浸润,管周组织内也有类似的细胞浸润
4.移植物抗宿主反应移植物抗宿主反应(graft versus host reaction,GVH)是免疫缺陷患者接受骨髓移植可发生的一种反应,。在此过程中,移植骨髓的部分干细胞分化成T细胞或B细胞,当其与宿主组织HLA相接触时可诱发:①CTL和淋巴因子形成,导致细胞介导免疫反应;②抗宿主HLA抗体形成,导致体液免疫反应,是GVH发生的主要机制。临床上,患者表现发热、体重减轻、剥脱性皮炎、肠吸收不良、肺炎及肝脾肿大等,其中些是由于感染所致。GVH的程度与供体和受体的HLA差别程度有关。本型反应多见于用骨髓移植治疗再生障碍性贫血、造血系统恶性肿瘤,特别是经细胞毒性药物或放射治疗后正常造血细胞和白血病细胞均被消灭的病例。
第三节 自身免疫性疾病
自身免病(autoimmune diseases)是指机体对自身抗原发生免疫反应而导致自身组织损害所引起的疾病。自从Donath 与Landsteiner提出此概念以来,许多疾病相继被列为自身免疫性疾病,值得提出的是,自身抗体的存在与自身免疫性疾病并非两个等同的概念,自身抗体可存在于无自身免疫性疾病的正常人特别是老年人,如抗甲状腺球蛋白、甲状腺上皮细胞、胃壁细胞、细胞核DNA抗体等。有时,受损或抗原性发生变化的组织可激发自身抗体的产生,如心肌缺血时,坏死的心肌可导致抗心肌自身抗体形成,但此抗本并无致病作用,是一种继发性免疫反应。因此,要确定自身免疫性疾病的存在一般需要根据:①有自身免疫反应的存在,②排除继发性免疫反应之可能,③排除其他病因的存在。
一、自身免疫性疾病的发病机制
自身免疫病的确切发病机制不明,可能与下列因素有关:
1.免疫耐受的丢失对特异性抗原不产生免疫应答的状态称免疫耐受。通常机体对自身抗原是耐受的,下列情况可导致失耐受:
(1)抗原性质变异:机体对于原本耐受的自身抗原,由于物理、化学药物、微生物等因素的是影响而发生变性、降解,暴露了新的抗原决定簇。例如变性的γ-球蛋白因暴露新的抗原决定簇而获得抗原性,从而诱发自身抗体(类风湿因子)。或通过修饰原本耐受抗原的截体部分,从而回避了TH细胞的耐受,导致免疫应答。这是由于大部分的自身抗原属于一种半抗原和载体的复合体,其中B细胞识别的是半抗原的决定簇,T细胞识别的是载体的决定簇,引起免疫应答时二种信号缺一不可,而一般机体对自身抗原的耐受性往往只是限于T细胞,如载体的抗原决定簇经过修饰,即可为T细胞识别,而具有对该抗原发生反应潜能的B细胞一旦获得TH的信号,就会分化、增殖,产生大量自身抗体。
(2)交叉免疫反应:与机体某些组织抗原成分相同的外来抗原称为共同抗原。由共同抗原刺激机体产生的共同抗体,可与有关组织发生交叉免疫反应,引起免疫损伤。例如A组B型溶血性链球菌细胞壁的M蛋白与人体心肌纤维的肌膜有共同抗原,链球菌感染后,抗链球菌抗体可与心肌纤维发生交叉反应,引起损害,导致风湿性心肌炎。
2.免疫反应调节异常TH细胞和T抑制细胞(TS)对自身反应性B细胞的调控作用十分重要,当Ts细胞功能过低或TH细胞功能过度时,则可有多量自身抗体形成。已知在NZB/WF1小鼠中随着鼠龄的增长Ts细胞明显减少,由于Ts细胞功能的过早降低,出现过量自身抗体,诱发与人类系统性红斑狼疮(SLE)类似的自身免疫性疾病。
3.遗传因素自身免疫病与遗传因素有较密切的关系,下列事实可说明这一情况:①很多自身免疫病如SLE、自身免疫性溶血性贫血、自身免疫性甲状腺炎等均具有家族史。②有些自身免疫病与HLA抗原表达的类型有联系,例如人类强直性脊柱炎与HLA-B27关系密切,已有报道将HLA-B27基因转至大鼠,转基因大鼠即可诱发强直性脊柱炎。
4.病毒因素病毒与自身免疫病的关系已在小鼠的自发性自身免疫病中得到一些证明,例如NZB小鼠的多种组织中有C型病毒及其抗原的存在,在病变肾小球沉积的免疫复合物中也有此类抗原的存在。病毒诱发自身免疫病的机制尚未完全清楚,可能是通过改变自身抗原载本的决定簇而回避了T细胞的耐受作用;也可能作为B细胞的佐剂(如EBV)促进自身抗体形成;或感染、灭活Ts细胞,使自身反应B细胞失去控制,产生大量自身抗体。此外,有些病毒基因可整合到宿主细胞的DNA中,从而引起体细胞变异(不能被识别)而引起自身免疫反应。
自身免疫性疾病往往具有以下共同特点:①患者有明显的家族倾向性,不少与HLA抗原尤其是与D/DR基因位点相关,女性多于男性;②血液中存在高滴度自身抗体和(或)能与自身组织成分起反应的致敏淋巴细胞;③疾病常呈现反复发作和慢性迁延的过程;④病因大多不明,少数由药物(免疫性溶血性贫血、血小板减少性紫癜)、外伤(交感性眼炎)等所致;⑤可在实验动物中复制出类似人类自身免疫病的模型。
二、自身免疫性疾病的类型和举例
自身免疫性疾病可分为二大类:
(一)器官特异性自身免疫病
组织器官的病理损害和功能障碍仅限于抗体或致敏淋巴细胞所针对的某一器官。主要有慢性淋巴性甲状腺炎、甲状腺功能亢进、胰岛素依赖型糖尿病、重症肌无力、慢性溃疡性结肠炎、恶性贫血伴慢性萎缩性胃炎、肺出血肾炎综合征(goodpasture syndrome)、寻常天疱疮、类天疱疮、原发性胆汁性肝硬变、多发性脑脊髓硬化症、急性特发性多神经炎等,其中常见者将分别于各系统疾病中叙述。
(二)系统性自身免疫病
由于抗原抗体复合物广泛沉积于血管壁等原因导致全身多器官损害,称系统性自身疫病。习惯上又称之为胶原病或结缔组织病,这是由于免疫损伤导致血管壁及间质的纤维素样坏死性炎及随后产生多器官的胶原纤维增生所致。事实上无论从超微结构及生化代谢看,胶原纤维大多并无原发性改变,以下简述几种常见的系统性自身免疫病。
1.系统性红斑狼疮(systemic lupus erythematosus, SLE) 是一种比较常见的系统性自身免疫病,具有以抗核抗体为主的多种自身抗体和广泛的小动脉病变及多系统的受累。临床表现主要有发热,皮损(如面部蝶形红斑)及关节、肾、肝、心浆膜等损害,以及全血细胞的减少。多见于年轻妇女,男女比为1:6~9,病程迁延反复,预后差。
病因与发病机制 本病的病因和发病机制不明,目前的研究主要集中在以下三个方面。
(1)免疫因素:患者体内有多种自身抗体形成,提示B细胞活动亢进是本病的发病基础。周围血中B细胞体外培养实验结果发现其增殖能力较正常强8~10倍。
(2)遗传因素:遗传因素与本病的关系表现为:①在纯合子双胎中有很高(69%)的一致性,②SLE患者家属成员中发病的可能性明显增加,③北美白人中SLe 与HLA DR2、DR3有关。这可能是由于位于HLA D区的免疫反应基因(Ir)对抗原(包括自身抗原)所激发的免疫反应的程度有调节作用的缘故。
(3)其他:非遗传因素在启动自身免疫反应中亦起着一定的作用。这些因素包括:①药物:盐酸肼苯哒嗪(hydralazine)、普鲁卡因酰胺(procainamide)等可引起SLE样反应。但停药后常可自愈;②病毒:在实验动物NZB和NZB/WF1小鼠中的自发性SLE样病中发现C型病毒感染,在肾小球中可检出病毒抗原-抗体复合物。但在SLE病中病毒因素尚未能充分得到证实;③性激素对SLE的发生有重要影响,其中雄激素似有保护作用,而雌激素则似有助长作用,故患者以女性为多,特别多发生在生育年龄,病情在月经和妊娠期加重。
自身抗体及组织损害机制本病患者体内有多种自身抗体,95%以上病人抗核抗体阳性,可出现抗DNA(双股、单股)、抗组蛋白、抗RNA-非组蛋白、抗核糖核蛋白(主要为Smith抗原)、抗粒细胞、抗血小板、抗平滑肌等抗体,其中抗双股DNA和抗Smith抗原具相对特异性,阳性率分别为60%和30%,而在其他结缔组织病的阳性率,均低于5%。
抗核抗体并无细胞毒性,但能攻击变性或胞膜受损的粒细胞,一旦它与细胞核接触,即可使胞核肿胀,呈均质状一片,并被挤出胞体,形成狼疮(LE)小体,LE小体对中性粒细胞、巨噬细胞有趋化性(图4-8),在补体存在时可促进细胞的吞噬作用。吞噬了LE小体的细胞为狼疮细胞(图4-9)。在组织中,LE小体呈圆或椭圆形,HE染色时苏木素着色而蓝染,故又称苏木素小体,主要见於肾小球或肾间质。一般仅在20%的患者可检见苏木素小体,为诊断SLE的特征性依据。
SLE的组织损害与自身抗体的存在有关,多数内脏病变是免疫复合物所介导(Ⅲ型变态反应),其中主要为DNA-抗DNA复合物所致的血管和肾小球病变,其次为特异性抗红细胞、粒细胞、血小板自身抗体经Ⅱ型变态反应导致相应血细胞的损害和溶解,引起全贫血。
病变急性坏死性小动脉、细动脉炎是本病的基础病变,几乎存在于所有患者并累及全身各器官。活动期病变以纤维素样坏死为主。慢性期血管壁纤维化明显,管腔狭窄,血管周围有淋巴细胞浸润伴水肿及基质增加。有时血管外膜纤维母细胞增生明显,胶原纤维增多,形成洋葱皮样结构,以脾中央动脉的变化最为突出。应用免疫组织化学方法可证实受累的血管壁中有免疫球蛋白、补体、纤维蛋白、DNA等存在,提示有抗原-抗体复合物机制的参与。
(1)肾:肾功能衰竭是SLE的主要死亡原因。SLE病人几乎均有不同程度的肾损害,约60%病便以狼疮性肾炎为主要表现.常见的类型有系膜增生型(10%~15%)、局灶增生型(10%~15%)、弥漫增生型(40%~50%)和膜型(10%~20%)。各型狼疮性肾炎的病变,类同于相应的原发性肾小球肾炎,各型病变间常有交叉,因此肾小球的病变呈多样性,晚期可出现典型的硬化性肾炎的表现。肾炎病变的发生主要基于肾小球中免疫复合物的沉积,可位于系膜区、内皮下和上皮下。其中弥漫增生型狼疮性肾炎中内皮下大量免疫复合物的沉积,是SLE急性期的特征性病变。在弥漫增生型及膜型病例中,约半数病例在间质及肾小管基膜上亦有免疫复合物沉积,因此肾小球病变和间质的炎症反应在狼疮性肾炎中十分明显(图4-10)。苏木素小体的出现有明确的诊断意义。

图4-8 红斑性狼疮花形细胞簇
游离的LE小体被多个粒细胞包围,形成花形细胞族(采自 Dubois)
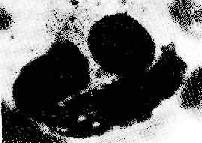
图4-9 狼疮细胞
胞浆内吞噬了两个LE小体,胞核被挤在一边
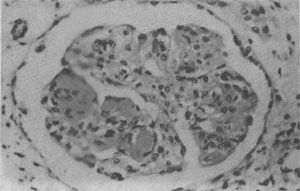
图4-10 狼疮性肾小球肾炎
肾小球毛细血管丛节段性纤维素样坏死,伴系膜细胞增生;间质炎细胞浸润
(2)皮肤:约40%的SLE病人有明显皮肤损害,以面部蝶形红斑最为典型,亦可累及躯干和四肢。镜下,表皮常有萎缩、角化过度、毛囊角质栓形成、基底细胞液化,表皮和真皮交界处水肿,基度膜、小动脉壁和真皮的胶原纤维可发生纤维素样坏死,血管周围常有淋巴细胞浸润。免疫荧光证实真皮与表皮交界处有IgG、IgM及C3的沉积,形成颗粒或团块状的荧光带即“狼疮带”,可能是坏死上皮细胞释出之抗原与血循环中弥散出来的抗核抗体等自身抗体形成的免疫复合物。狼疮带的出现对本病有诊断意义。
(3)心:大约半数病例有心脏受累,心瓣膜非细菌性疣赘性心内膜炎(nonbacterial verrucous endocarditis或Libman-Sach endocarditis)最为典型,敖生物常累及二尖瓣或三尖瓣,其特点为:大小自1mm至3~4mm,数目单个或多个不等,分布极不规则,可累及瓣膜之前后面或心腔之内膜或腱索(图4-11)。镜下,赘生物由纤维蛋白和坏死碎屑及炎症细胞构成,根部基质发生纤维素样坏死,伴炎细胞浸润,后期发生机化。
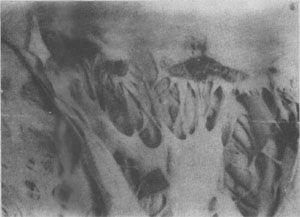
图4-11 红斑性狼疮的心瓣膜疣状心内膜炎
二尖瓣上有疣状赘生物形成
(4)关节:90%以上的病例有不同程度的关节受累。滑膜充血水肿,有较多单个核细胞浸润。于紧接上皮浅表部位的结缔组织可出现灶性纤维素样坏死,但很少侵犯关节软骨等深部组织,因此极少引起关节畸形。
(5)肝:约25%的病例可出现肝损害,称狼疮性肝炎。可表现为汇管区及汇管区周围的单个核细胞浸润及附近肝细胞的碎屑状坏死等慢性活动性肝炎的典型病变。亦可仅有少量散在分布的小灶性坏死等轻微病变。
(6)脾:体积略增大,包膜增厚,滤泡增生颇常见。红髓中有多量浆细胞,内含IgG、IgM,最突出的变化是小动脉周围纤维化,形成洋葱皮样结构(图4-12)。
图4-12 红斑性狼疮之脾病变
脾小体中央细动脉壁呈洋葱皮样结构
(7)淋巴结:全身淋巴结均有不同程度的肿大,窦内皮增生。其中较多的浆细胞,小血管变化与脾所见相同。
2.口眼干燥综合征口眼干燥综合症(Sjǒgren syndrome)临床上表现为眼干、口干等特征,乃唾液腺、泪腺受免疫损伤所致。本病可单独存在,也可与其他自身免疫病同时存在,后者最常见的是类风湿性关节炎、SLE等。病变主要累及唾液腺及泪腺,其他外分泌腺包括呼吸道、消化道腺体也可受累。
唾液腺的组织学病变主要表现为腺管周围大量炎细胞浸润,主要是淋巴细胞和浆细胞,有时可形成淋巴滤泡并有生发中心形成。伴腺管上皮增生,引起管腔阻塞。病变晚期腺泡萎缩、纤维化,为脂肪组织所替代。个别病例浸润的淋巴细胞形成淋巴瘤样结构。由于唾液腺的破坏而引起口腔粘膜干裂及溃疡形成。
泪腺的类似病变可导致角膜上皮干燥、炎症及溃疡形成。呼吸道、消化道受累可导致相应的鼻炎、喉炎、支气管炎、肺炎及萎缩性胃炎。在肾可发生间质性肾炎,肾小管周围大量单个核细胞浸润,导致肾小管萎缩、纤维化,因肾小管功能损害而引起肾小管性酸中毒、磷酸盐尿等颇常见。
淋巴结肿大并有增生性变化,核分裂多,故又名假性淋巴瘤。值得提出的是本病患者发生恶生淋巴瘤的机会较正常人高40倍。
发病机制 本病的发病机制尚不清楚。由于常伴发SLE和类风湿性关节炎,提示本病的发生与免疫性损伤有关。患者B细胞功能过度,表现为多克隆高球蛋白血症和类风湿因子(RF)、抗核抗体、冷球蛋白及抗唾液腺抗体的形成。近年来并发现两种特征性抗核糖核蛋白成分的自身抗体,分别命名为抗SS-B和SS-A,在本病有很高的阳性率(60%、70%),对本病的诊断有参考价值。病灶处有大量B及T细胞浸润,后者大部分为T辅助细胞,也有一部分为T杀伤细胞,提示亦有细胞免疫机制的参与。
3.类风湿性关节炎详见骨关节疾病章。
4.硬皮病硬皮病(scleroderma)又名进行性系统性硬化症(progressive systemic sclerosis),以全身许多器官间质过度纤维化为其特征。95%以上的患者均有皮肤受累的表现;但横纹肌及许多器官(消化道、肺、肾、心等)受累是本病主要损害所在,病变严重者可导致器官功能衰竭,威胁生命。
病因和发病机制本病病因不明,其发病可能与以下因素有关:
(1)胶原合成增加:体外培养证实,患者纤维母细胞合成胶原的能力明显高于正常人,合成超过降解,导致大量胶原纤维的积集;
(2)Ⅳ型变态反应:在皮肤病变中有T细胞浸润,所分泌的淋巴因子及其刺激巨噬细胞分泌的因子可刺激纤维母细胞大量合成胶原;
(3)自身抗体:50%患者有轻度高丙种球蛋白血症及多种自身抗体,包括RF,抗平滑肌抗体,抗核抗体等,可能由于抗原抗体免疫复合物的沉积或内皮细胞毒的作用,造成小血管内皮细胞损伤、血栓形成、管壁纤维化、管腔狭窄,导致组织缺氧而引起纤维间质增生。
【病变】
(1)皮肤:病变由指端开始,向心性发展,累及前臂、肩、颈、脸,使关节活动受限。早期受累的皮肤发生水肿,质韧。镜下,主要表现为小血管周围淋巴细胞浸润,毛细血管内皮细胞肿胀、基膜增厚、管腔部分阻塞,间质水肿,胶原纤维肿胀,嗜酸性增强。随着病变的发展,真皮中胶原纤维明显增加,并与皮下组织紧密结合,表皮萎缩变平,黑色素增加,钉突和附属器萎缩消失,小血管增厚、玻璃样变。晚期手指细而呈爪状、关节活动受限,有时指端坏死甚或脱落,面部无表情呈假面具状。
(2)消化道:约有1/2患者消化道受累,粘膜上皮萎缩,固有层、粘膜下层、肌层为大量胶原纤维所取代,血管周单个核细胞浸润。病变以食管下2/3段最严重,管腔狭窄,缺乏弹性。小肠、结肠也可受累。临床上出现吞咽困难、消化不良等症状。
(3)肾:叶间小动脉病变最为突出,表现为内膜粘液样变性,伴内皮细胞增生及随后的管壁纤维化、管腔明显狭窄,部分病例并有细动脉纤维素样坏死。临床上可出现高血压,与恶性高血压肾病变难以区别。约50%患者死于肾功能衰竭。
(4)肺:弥漫性间质纤维化,肺泡扩张、胞泡隔断裂,形成囊样空腔,本病是造成蜂窝肺的重要原因之一。
5.结节性多动脉炎结节性多动脉炎(polyarteritis nodosa)是全身动脉系统的疾病,表现为中小动脉壁的坏死性炎症。患者以青年人为多,有时也可发生在儿童及老人、男女之比为2~3:1。
病变各系统或器官的中小动脉均可受累,其中以肾(85%)、心(75%)、肝(65%)、消化道(50%)最为常见。此外,胰、睾丸、骨骼肌、神经系统和皮肤也可受累。
病变多呈节段性,以血管分叉处最为常见。内眼观,病灶处形成直径约2~4mm的灰白色小结节,结节之间的血管壁外观正常。镜下,急性期表现为急性坏死炎症,病变从内膜和中膜内层开始,扩展至管壁全层及外膜周围,纤维素样坏死颇为显著,伴炎细胞浸润(图4-13)尤以嗜酸性及中性粒细胞为多,继而有血栓形成。以后的进展是纤维增生,管壁呈结节性增厚,管腔机化阻塞和明显的动脉周围纤维化。值得注意的是早期炎性坏死变化及后期胶原化可同时存在。病变的主要后果是缺血性损害和梗死形成。
图4-13 结节性多动脉炎
两个动脉壁的各层都有炎性细胞浸润,外膜尤为显著。中膜发生纤维素样坏死
本病病变分布广泛,临床表现变异多端,患者常有低热、乏力、粒细胞增多以及多系统受累的症状,如血尿、肾功能衰竭、高血压、腹痛、腹泻、黑粪及周围神经炎等。病程快慢不一,经免疫抑制治疗,55%患者可存活。
病因与发病机制 病因和发病机制不明,动物实验提示,体液因素在本病的发生中起着重要作用。免疫荧光技术证实,人结节性多动脉炎血管壁中有免疫球蛋白和补体,有些还有HBsAg,约50%患者血清HBsAg或抗HBs阳性。
6.Wegener肉芽肿病Wegener肉芽肿病是一种少见病,具有以下特点:①小血管急性坏死性脉管炎,可累有各器官的血管,以呼吸道、肾、脾最常受累。表现为小动脉、小静脉管壁的纤维素样坏死,伴弥漫性中性和嗜酸性粒细胞浸润;②呼吸道肉芽肿性坏死性病变,可累及口、鼻腔、鼻旁窦、喉、气管、支气管和肺。病变为由大量积集的单核巨噬细胞、淋巴细胞以及少量多核巨细胞、类上皮细胞、纤维母细胞组成的肉芽肿,中央可陷于成片凝固性坏死。肉眼常形成明显的肿块,表面则因坏死溃破而有溃疡形成;③坏死性肾小球肾炎,表现为在局灶性或弥漫增生性肾小球肾炎的基础上,有节段性毛细血管袢的纤维素样坏死,血栓形成,如未经治疗可发展为快速进行性肾炎,病程凶险,出现进行性肾功能衰竭。
本病的病因不明,由于有明显的血管炎,并于局部可检得免疫球蛋白和补体,提示其发病与Ⅲ型变态反应有关。但呼吸道出现的肉芽肿和坏死性病变,又提示可能与Ⅳ型变态反应有关,临床上应用细胞毒药物大多能使本病缓解。
第四节 免疫缺陷病
免疫缺陷病(immunodeficiency diseases)是一组由于免疫系统发育不全或遭受损害所致的免疫功能缺陷引起的疾病。有二种类型:①原发性免疫缺陷病,又称先天性免疫缺陷病,与遗传有关,多发生在婴幼儿。②继发性免疫缺陷病,又称获得性免疫缺陷病,可发生在任何年龄,多因严重感染,尤其是直接侵犯免疫系统的感染、恶性肿瘤、应用免疫抑制剂、放射治疗和化疗等原因引起。
免疫缺陷病的临床表现因其性质不同而异,体液免疫缺陷的患者产生抗体的能力低下,因而发生连绵不断的细菌感染。淋巴组织中无生发中心,也无浆细胞存在。血清免疫球蛋白定量测定有助于这类疾病的诊断。细胞免疫缺陷在临床上可表现为严重的病毒、真菌、胞内寄生菌(如结核杆菌等)及某些原虫的感染。患者的淋巴结、脾及扁桃体等淋巴样组织发育不良或萎缩,胸腺依赖区和周围血中淋巴细胞减少,功能下降,迟发性变态反应微弱或缺如。免疫缺陷患者除表现难以控制的感染外,自身免疫病及恶性肿瘤的发病率也明显增高。
一、原发性免疫缺陷病
原发性免疫缺陷病是一组少见病,与遗传相关,常发生在婴幼儿,出现反复感染,严重威胁生命。因其中有些可能获得有效的治疗,故及时诊断仍很重要。按免疫缺陷性质的不同,可分为体液免疫缺陷为主、细胞免疫缺陷为主以及两者兼有的联合性免疫缺陷三大类。此外,补体缺陷、吞噬细胞缺陷等非特异性免疫缺陷也属于本组。我国各类原发性免疫缺陷病的确切发病率尚不清楚,其相对发病百分率大致为体液免疫缺陷占50%,细胞免疫缺陷10%,联合免疫缺陷30%,吞噬细胞功能缺陷6%,补体缺陷4%。
1.体液免疫(B细胞)缺陷为主的疾病表现为免球蛋白的减少或缺乏,包括:
(1)原发性丙种球蛋白缺乏症:有两种类型:①Bruton型,较常见,为婴儿性联丙种球蛋白缺乏病,与X染色体隐性遗传有关,仅发生于男孩,于出生半年以后开始发病;②常染色体隐性遗传型,男女均可受累,也可见于年人。本病的特点在于:血中B细胞明显减少甚至缺如,血清免疫球蛋白(IgM、IgG、IgA)减少或缺乏,骨髓中前B细胞发育停滞。全身淋巴结、扁桃体等淋巴组织生发中心发育不全或呈原始状态;脾和淋巴结的非胸腺依赖区淋巴细胞稀少;全身各处浆细胞缺如。T细胞系统及细胞免疫反应正常。
由于免疫缺陷,患儿常发生反复细菌感染,特别易受流感嗜血杆菌、酿脓链球菌、金黄色葡萄球菌、肺炎球菌等感染,可引起中耳炎、鼻窦炎、支气管炎、肺炎、脑膜炎或败血症而致死。注射丙种球蛋白,能控制感染,但由于无法提高呼吸道等粘膜处的SlgA,因此鼻部、肺部的感染极易复发。
(2)孤立性IgA缺乏症:本病是最常见的先天性免疫缺陷病,患者的血清IgA和粘膜表面分泌型IgA(SlgA)均缺乏。可以是家族性或获得性,前者通过常染色体隐性或显性遗传。患者多无症状,有些可有反复鼻窦或肺部感染及慢性腹泻、哮喘等表现。自身免疫、过敏性疾病的发病率也较高。血清Iga 低下(<5mg/dl)为确诊本病的重要依据。
本病的发病与IgA B细胞的分化障碍有关。患者Iga B细胞的数量正常,但多数为不成熟表型,在体外仅少数能转化为IgA细胞。
约有50%的本病患者血清中含IgA自身抗体。因此应避免注射含IgA的血制品,如错误地给予IgA或输血治疗,可引起过敏性休克。
(3)普通易变免疫缺陷病:普通易变免疫缺陷病(common variable immunodeficiency)是相当常见而未明确了解的一组综合征。男女均可受累,发病年龄在15~35岁不等,可为先天性或获得性。其免疫缺陷累及范围可随病期而变化,起病时表现为低丙种球蛋白血症,随着病情进展可并发细胞免疫缺陷。其特点是:①低丙种球蛋白血症,免疫球蛋白总量和IgG均减少;②2/3患者血循环中B细胞数量正常,但不能分化为浆细胞;淋巴结、脾、消化管淋巴组织中B细胞增生明显,但缺乏浆细胞。部分病例有T辅助细胞减少、T抑制细胞过多;部分病例有抗T细胞B细胞的自身抗体;或巨噬细胞功能障碍;③患者主要表现为呼吸道、消化道的持续慢性炎症,自身免疫病的发病率也较高。
2.细胞免疫(T细胞)缺陷为主的疾病单纯T细胞免疫缺陷较为少见,一般常同时伴有不同程度的体液免疫缺陷,这是由于正常抗体形成需要T、B细胞的协作。T细胞免疫缺陷病的发生与胸腺发育不良有关,故又称胸腺发育不良或Di George综合征。本病与胚胎期第Ⅲ、Ⅳ对咽囊发育缺陷有关,因此,患者常同时有胸腺和甲状旁腺缺如或发育不全,先天性心血管异常(主动脉缩窄、主动脉弓右位畸形等)和其他脸、耳畸形。周围血循环中T细胞减少或缺乏,淋巴组织中浆细胞数量正常,但皮质旁胸腺依赖区及脾细动脉鞘周围淋巴细胞明显减少。常在出生后即发病,主要表现为各种严重的病毒或真菌感染,呈反复慢性经过。近年来应用胸腺素(thymosin)或胚胎胸腺上皮移植治疗,获得一定疗效。
3.联合性免疫缺陷病
(1)重症联合性免疫缺陷病:本病是一种体液免疫、细胞免疫同时有严重缺陷的疾病,一般T细胞免疫缺陷更为突出。患者血循环中淋巴细胞数明显减少,成熟的T细胞缺如,可出现少数表达CD2抗原的幼稚的T细胞。免疫功能如,无同种异体排斥反应和迟发型过敏反应,也无抗体形成。本病的基本缺陷尚不清楚,可能与干细胞分化为T、B细胞发生障碍或胸腺及法氏囊相应结构的发育异常有关。病变主要表现为淋巴结、扁桃体及阑尾中淋巴组织不发育;胸腺停留在6~8周胎儿的状态,其中无淋巴细胞或胸腺小体,血管细小。患儿由于存在体液和细胞免疫的联合缺陷,对各种病原生物都易感,临床上常发生反复肺部感染、口腔念珠菌感染、慢性腹泻、败血症等。在治疗方面可选用正常骨髓干细胞移植或同胞兄妹骨髓移植。但供体骨髓中T细胞介导的移埴物抗宿主反应(GVH)往往是造成治疗失败的严重问题。
约有25%~50%的重症联合免疫缺陷病例,主要与先天性缺乏腺苷脱氨酶(adenosine deaminase,ADA)有关。一则严重影响细胞DNA包括淋巴细胞DNA的合成代谢,同时由于三磷酸脱氧腺苷在淋巴细胞的堆积,后者对淋巴细胞尤其是T细胞具有一定的毒性作用,从而造成淋巴细胞在增补、分化及功能方面的障碍。测定病人红细胞的ADA有助于诊断本病。给病儿输入含有ADA活性的红细胞可一定程度地改善其免疫功能状态。
(2)伴血小板减少和湿疹的免疫缺陷病:本病又称Wiscott-Aidrich综合征,是一种X染色体隐性遗传性免疫缺陷病,多见于男孩,临床表现为湿疹、血小板减少及反复感染。免疫缺陷早期表现为对多糖类抗原的体液免疫应答不全,患儿对肺炎球菌和其他带多糖荚膜的细菌特别易感。随着年龄增长,逐渐出现细胞免疫缺陷,易患病毒和卡氏肺孢子虫感染。因血小板功能下降而常伴明显的出血倾向。血中IgM明显减少,IgG正常,在有些患者IgA、IgE可升高,淋巴细胞数通常正常。各种疾苗接种后,抗体形成反应微弱,T细胞功能欠佳,迟发型变态反应不良,患者中恶性淋巴瘤发病率较高。骨髓移植对有些患有一定疗效。
(3)伴共济失调和毛细血管扩张症的免疫缺陷病:本病是常染色体隐性遗传性疾病,常累及幼儿,兼有T、B细胞免疫缺陷。一般2岁开始起病,临床特点包括小脑性共济失调,眼结膜和皮肤毛细血管扩张,反复鼻窦及肺部感染等。过去认为本病是一种神经系统疾病,目前已知除神经系统外,血管、内分泌及免疫系统均可受累,40%患者显示选择性IgA缺陷。
患者胸腺发育不良,淋巴细胞和胸腺小体均严重缺乏,皮质髓质界限模糊。淋巴结无滤泡形成,浆细胞也少见。关于本病出现多系统异常的机制,尚无一致的见解,可能与DNA修复功能障碍有关,患者并发恶性淋巴瘤的机率甚高。
4.吞噬细胞功能障碍本病表现为吞噬细胞数量减少、游走功能障碍、吞噬能力虽正常,但由于胞内缺乏各种消化病原的酶而丧失了杀灭和消化病原的能力。患者对致病与非致病微生物均易感,因而易发生反复感染,其中慢性肉芽肿病是一种X染色体隐性遗传性疾病,一般在2岁左右起病,表现为颈淋巴结、皮肤、肺、骨髓等处慢性化脓性炎或肉芽肿性炎、肝、脾肿大。
5.补体缺陷补体在炎症及免疫反应中起着重要作用,常见的补体缺陷有①C3缺乏或C3抑制物缺乏,后者使C3过度消耗同样使血清中C3水平下降,导致反复细菌感染。②C1抑制物缺陷,C1抑制物是血清中的一种糖蛋白,除对Cis有抑制作用外,尚可抑制纤溶酶原、激肽等炎症介质的激活,因此C1抑制物的缺陷,可导致血管通透性增加、组织水肿,即所谓的遗传性血管水肿。
二、 继发性免疫缺陷病
许多疾病可伴发继发性免疫缺陷病,包括感染(风疹、麻疹、麻风、结核病、巨细胞病毒感染、球孢子菌感染等)、恶性肿瘤(何杰金病、急性及慢性白血病、骨髓瘤等)、自身免疫性疾病(SLE、类风湿性关节炎等)、蛋白丧失(肾病综合征、蛋白丧失肠病)、免疫球蛋白合成不足、淋巴细胞丢失(因药物、系统感染等)以及某些其他疾病(如糖尿病、肝硬变、亚急性硬化性全脑炎)和免疫抑制治疗等。继发性免疫缺陷病可以是暂时性的,当原发疾病得到治疗后,免疫缺陷可恢复正常;也可以是持久性的。继发性免疫缺陷常由多因素参与引起,例如癌肿伴发的继发性免疫缺陷病可由于肿瘤、抗癌治疗和营养不良等因素所致。
继发性免疫缺陷病较原发性者更为常见,但无特征性的病理变化。本病的重要性在于机会性感染所引起的严重后果。因此及时的诊断和治疗十分重要。本节着重途述发病率日增而死亡率极高的获得性免疫缺陷综合征(艾滋病)。
获得性免疫缺陷综合征(acquired immunodeficiency syndrome,AIDS)自1981年首先由美国疾病控制中心报道以来,不仅在美国而且在全球日益蔓延。根据1993年8月世界卫生组织的调查估计,目前世界上已有1400万人携带HIV,死亡人数达200万,其中70%是中非和东非人。本病的特点为T细胞免疫缺陷伴机会性感染和(或)继发性肿瘤。临床表现为发热、乏力、体重下降、腹泻、全身淋巴结肿大及神经系统症状。50%患者有肺部机会性卡氏肺孢子虫(pneumocystis carinii)感染,其他机会性病原有曲霉、白色念珠菌、隐球菌,巨细胞病毒(cytomegalovirus)、疱疹病毒(herpes virus )和弓形虫(toxoplasma)等。此外,约有1/3患者有多发性Kaposi肉瘤、淋巴瘤等。病情险恶,死亡率高。流行病学调查发现,本病的发生与以下危险因素有关:①男性同性恋,约占70%;②静脉注射毒品约占17%;③接受血制品而获得感染者约占1%;④双亲都具有上述危险因素的婴儿以及与高危险人有异性性接触者,等等。
【病因和发病机制】
本病的病因是人类免疫缺陷病毒(HIV)。许多事实证实该病毒与AIDS的关系密切,表现为:在患者的淋巴样细胞和体液(精液、唾液及脑脊液)中可分离出HIV;抗HIV抗体几乎在90%的患者中可被检出,与正常人群中1%的检出率相比有显著的差别;此外,与HIV有交叉反应的STLV-Ⅲ在猕猴中能引起AIDS样病。
AIDS病的主要传染途径为:①性交接触感染,最为常见;②应用污染的针头作静脉注射;③输血和血制品的应用;④母体病毒经胎盘感染胎儿或通过哺乳、粘膜接触等方式感染婴儿。
HIV选择性地侵犯和破坏TH细胞是AIDS发病的症结所在,由于HIV与TH细胞表面的CD4分子高度亲和,因而CD4分子可认为是HIV的受体乃入侵门户。HIV为一种C型逆转录病毒,分核心及包壳二大部分,包壳的糖蛋白gp120和ap41可先后与TH细胞膜上的CD4分子结合而进入TH细胞。在细胞内,病毒基因经逆转录而产生前病毒DNA,后者一经整合到宿主细胞的DNA,即可转录出完整的病毒颗粒,大量病毒颗粒在CD4+细胞膜处通过出芽而释放,并导致该细胞的溶解和死亡。由于TH细胞是调节整个免疫系统的枢纽细胞,TH细胞的消减必然影响到IL-2、γ-干扰素以及激活巨噬细胞、B细胞等有关的多种淋巴因子的分泌,将进一步影响TH细胞及其他免疫活性细胞的功能,包括:①TH细胞克隆增生和混合淋巴细胞反应降低,淋巴因子减少,对可溶性抗原的反应也减弱;②Ts(Tc)细胞克隆增生降低,特异性细胞毒反应减少;③NK细胞杀灭肿瘤细胞的功能降低;④B细胞在特异性抗原刺激下不产生正常的抗体反应,而原因不明的激活和分化引起高丙种球蛋白血症;⑤巨噬细胞对一般信号无反应,溶解肿瘤细胞、杀灭胞内寄生菌、真菌、原虫的功能减弱(图4-14)。
近年来发现HIV尚可感染组织中的单核巨噬细胞。由于巨噬细胞表面亦有少量CD4分子存在,因此其感染方式可通过HIV的gp120与CD4分子结合;更主要是通过抗HIV的凋理,经巨噬细胞的Fc受体吞噬HIV而使细胞受染。在巨噬细胞内复制的病毒,通常贮藏于胞浆内,不像TH细胞那样在胞膜上大量出芽,因而巨噬细胞不会迅速死亡,反可作为“地下工厂”及运载工具将病毒运至其他部位。例如已经发现,在脑组织,以小胶质细胞内含的HIV最多,但可能来自巨噬细胞的运送。受HIV感染后的巨噬细胞,其递呈抗原及分泌单核因子等功能亦将受到抑制。
综合以上后果,导致严重免疫缺陷,构成了AIDS发病的中心环节。此外遗传素质对本病的发生也可能有一定影响,ADIS患者中HLA-DR5抗原阳性率较高。
图4-14 HIV侵犯TH细胞后免疫调节障碍示意图
【病变】
病变可归纳为全身淋巴样组织的变化,机会性感染和恶性肿瘤三个方面。
1.淋巴样组织的变化早期及中期,淋巴结肿大。镜下,最初有淋巴滤泡明显增生,生发中心活跃,髓质出现较多浆细胞。随后滤泡的外套层淋巴细胞减少或消失,小血管增生,并有纤维蛋白样物质或玻璃样物质沉积,生发中心被零落分割。副皮质区的淋巴细胞(CD4+细胞)进行性减少,代之以浆细胞浸润。晚期的淋巴结病变,往往尸检时才能看到,呈现一片荒芜,淋巴细胞,包括T、B细胞几乎均消失殆尽,无淋巴滤泡及副皮质区之分,仅有一些巨噬细胞和浆细胞残留(图4-15)。有时特殊染色可显现大量分枝杆菌、真菌等病原微生物,却很少见到肉芽肿形成等细胞免疫反应性病变。
AIDS病人的脾呈轻度肿大,镜下有淤血,T、B细胞均减少,淋巴滤泡及淋巴鞘缺如。死于感染的病例,脾内常有较多中性粒细胞及一些吞噬病原微生物的巨噬细胞。扁桃体、小肠、阑尾和结肠内的淋巴样组织均萎缩,淋巴细胞明显减少。胸腺的组织与同龄人相比,呈现过早萎缩,淋巴细胞减少、胸腺小体钙化。
2.继发性感染多发性机会感染是本病另一特点,感染的范围广泛,可累及各器官,其中以中枢神经系统、肺、消化道的疾病最为常见。病原种类繁多,一般可有二种以上感染同时存在。由于严重的免疫缺陷,感染所致之炎症反应往往轻而不典型。如肺部结核菌感染,很少形成典型的肉芽肿性病变,而病灶中的结核杆菌却甚多。
约有半数病例有卡氏肺孢子虫感染,因之对诊断本病有一定参考价值。病变区肺间质及肺泡腔内有较多巨噬细胞及浆细胞浸润,其特征性变化是肺泡腔内出现由大量免疫球蛋白及原虫组成的伊红色泡沫样渗出物。
约70%的病例有中枢神经系统受累,其中继发性机会感染有播散性弓形虫或新型隐球菌(cryptococcus neoformans)感染所致的脑炎或脑膜炎;巨细胞病毒和乳多空病毒(papovavirus)所致的进行性多灶性白质脑病等。由HIV直接引起的疾病有脑膜炎、亚急性脑病、痴呆等。这一情况提示,除淋巴细胞、巨噬细胞外,神经系统也是HIV感染的靶组织。
3.恶性肿瘤 约有30%患者可发生Kaposi肉瘤。该肿瘤为血管内皮起源,广泛累及皮肤、粘膜及内脏,以下肢最为多见。肉眼观肿瘤呈暗蓝色或紫棕色结节。镜下显示成片梭形肿瘤细胞,构成毛细血管样空隙,其中可见红细胞。与典型的Kaposi肉瘤不同之处在于其多灶性生长和进行性临床过程。其他常见的伴发肿瘤包括未分化性非何杰金淋巴瘤、何杰金和Burkitt淋巴瘤等,脑原发性淋巴瘤也很常见。
【临床病理联系】
临床上将AIDS的病程分为三个阶段:①早期或称急性期,感染病毒3~6周后可出现咽痛、发热、肌肉酸痛等一些非特异性表现。病毒在体内复制,但由于患者尚有较好的免疫反应能力,2~3周后这种急性感染症状可自行缓解;②中期或称慢性期,机体的免疫功能与病毒之间处于相互抗衡阶段,在某些病例此期可长达数年或不再进入末期。此期病毒复制持续于低水平,临床可以无明显症状或出现明显的全身淋巴结肿大,常伴发热、乏力、皮疹等;③后期或称危险期,机体免疫功能全面崩溃,病人持续发热、乏力、消瘦、腹泻,并出现神经系统症状,明显的机会感染及恶性肿瘤,血液化验可见淋巴细胞明显减少(<30%),CD4+细胞减少尤为显著,CD4+细胞与CD8+细胞之比可由原来的2下降至0.5以下,细胞免疫反应丧失殆尽。
本病的预后差,死亡率达100%,致病原因虽已清楚,但制备有效的疫苗尚有待时日,其困难在于HIV在不同的患者有惊人的多型性,目前又无理想的治疗药物,因此大力开展预防,对防止AIDS流行至关重要。
图4-15 AIDS淋巴结
淋巴细胞明显减少,无淋巴滤泡及副皮质区之分
第五章 炎症
外源性和内源性损伤因子可引起细胞各种各样的损伤性病变,与此同时机体的局部和全身则发生一系列复杂的反应,以局限和消灭损伤因子,清除和吸收坏死组织、细胞,并修复损伤,这就是机体的防御性反应—炎症(inflammation)。
第一节 概述
一、炎症的概念
1.炎症的定义具有血管系统的活体组织对损伤因子所发生的防御反应为炎症。虽然单细胞动物和其它无血管的多细胞动物对损伤因子也可发生反应,例如吞噬反应或其它清除有害因子的反应,但这些都不能称为炎症。只有当进化到具有血管的机体,才具有以血管反应为主要特征,同时又保留上述吞噬和清除等反应的复杂而又完善的炎症过程。因此,从进化角度看,血管反应是炎症过程的中心环节。
在炎症过程中,一方面损伤因子直接和间接造成组织和细胞的破坏,另一方面通过炎症充血和渗出反应,以稀释、杀伤和包围损伤因子。同时通过实质和间质细胞的再生使受损伤的组织得以修复和愈合。因此可以说炎症是损伤的抗损伤的统一过程。
2.炎症的局部表现和全身反应炎症的局部临床特征是红、肿、痛和功能障碍。红、热是由于炎症局部血管扩张、血流加快所致。肿是由于局部炎症性充血、血液成分渗出引起。由于渗出物压迫和某些炎症介质直接作用于神经末梢而引起疼痛。基于炎症的部位、性质和严重程度将引起不同的功能障碍,如肺炎影响气血交换从而引起缺氧和呼吸困难等。
炎症所引起的全身反应包括发热和末梢血白细胞计数增多。发热在感染性炎症,特别是当病原体蔓延入血时常表现很突出。白细胞所产生的白细胞介素1和肿瘤坏死因子,以及前列腺素E均可引起交感神经兴奋,使皮肤血管收缩,散热降低而造成发热。在细菌感染引起的炎症时,末梢血白细胞计数可达15×109~20×109/L,甚至更高,这主要是由于白细胞介素1和肿瘤坏死因子所引起的骨髓中白细胞储存库释放加速,而且相对不成熟的杆状核中性粒细胞所占比例增加,这就是临床上所称的“核左移”。持续稍久的感染还可以通过集落刺激因子的产生而促进骨髓造血前体细胞的增殖。某些病毒性疾病和伤寒等炎症还能出现末梢血白细胞计数降低。
3.炎症反应的防御作用以血管系统改变为中心的一系列局部反应,有利于清除消灭致病因子,液体的渗出可稀释毒素,吞噬搬运坏死组织以利于再生和修复,使致病因子局限在炎症部位而不致蔓延全身。因此,炎症是机体的防御性反应,通常对机体是有利的,如果没有炎症反应,人们将不能长期生存于这个充满致炎因子的自然环境中。
但是炎症对机体也有潜在的很大危害性,严重的过敏反应可危及病人的生命;心包腔内纤维素性渗出物机化可形成缩窄性心包炎,进而影响心功能;发生于脑实质或脑膜的炎症可引起颅内压升高,甚至形成脑疝致使生命中枢受压而造成病人死亡;此外声带急性炎症水肿可导致窒息,等等。因此在一定情况下应采取措施控制炎症反应。
二、炎症的原因
任何能够引起组织损伤的因素都可成为炎症的原因。虽然致炎因子种类繁多,但可归纳为以下几大类。
1.物理性因子高热、低温、放射线及紫外线等。
2.化学性因子包括外源性和内源性化学物质。外源性化学物质有强酸、强碱等腐蚀性物质及松节油、芥子气等。内源性化学毒物如坏死组织的分解产物及在某些病理条件下堆积于体内的代谢产物如尿素等。
3.机械性因子如切割、撞击、挤压等。
4.生物性因子细菌、病毒、立克次体、支原体、真菌、螺旋体和寄生虫等为炎症最常见的原因。它们通过在体内繁殖,产生、释放毒素直接导致细胞和组织损伤,而且还可通过其抗原性诱发免疫反应导致炎症。
5.免疫反应各型变态反应均能造成组织和细胞损伤而导致炎症:Ⅰ型变态反应如过敏性鼻炎、荨麻疹;Ⅱ型变态反应如抗基底膜性肾小球肾炎;Ⅲ型变态反在如免疫复合物性肾小球肾炎和Ⅳ型变态反应如结核、伤寒等等;此外还有某些自身免疫性疾病如淋巴性甲状腺炎、溃疡性结肠炎等。
损伤因子作用于机体是否引起炎症,以及炎症反应的强弱不仅与损伤因子的性质和损伤的强度有关,并且还与机体对损伤因子的敏感性有关,如幼儿和老年人免疫功能低下,易患肺炎,病情也较严重;接种过预防疫苗的儿童,对该病原体常表现不感受性等。因此,炎症反应的发生和发展取决于损伤因子和机体反应性两方面的综合作用。
三、炎症的基本病理变化
炎症的基本病理变化包括局部组织的变质(alteration)、渗出(exudation)和增生(proliferation)。在炎症过程中这此病理变化按照一定的先后顺序发生,一般早期以变质和渗出变化为主。后期以增生为主,但三者是相互密切联系的。一般地说,变质属于损伤过程,而渗出和增生则属于抗损伤过程。
1.变质炎症局部组织发生的变性和坏死称为变质。变质既可发生于实质细胞,也可见于间质细胞。实质细胞常出现的变质包括细胞水肿、脂肪变性、凝固性或液化性坏死等。间质结缔组织的变质可表现为粘液变性,纤维素样变性或坏死等。
致炎因子的直接损伤作用、炎症过程中发生的血液循环障碍和炎症反应产物的共同作用,造成局部组织的变质。因此变质的轻重是由致炎因子和机体反应两个方面决定的。
2.渗出炎症局部组织血管内的液体、蛋白质和血细胞通过血管壁进入间质、休腔、体表或粘膜表面的过程称为渗出。以血管反应为中心的渗出性病变是炎症的重要标志,在局部具有重要的防御作用。
炎症渗出是由于局部血管通透性升高和白细胞为主动游出所致。在炎症渗出液(exudate)内蛋白质含量较高,并含有较多的细胞成分和其碎屑。因而由炎症所引起的浆膜腔渗出液,比重高于1.020,外观混蚀,细胞含量多,这些有别于单纯因静脉回流受阻所形成的漏出液(transudate)。渗出液和漏出液均可在组织间质聚积,造成水肿(edema),或在浆膜腔造成积液(hydrops)。
3.增生在致炎因子、组织崩解产物或某些理化因子刺激下,炎症局部的巨噬细胞、内皮细胞和纤维母细胞可增生。在某些情况下局部的上皮细胞或实质细胞也可增生。正是这种增生反应使损伤组织得以修复。许多生长因子参与刺激间质和实质细胞的增生,其机制与再生和修复过程相似。
第二节 急性炎症
炎症通常可依病程经过分为两大类:急性炎症(acute inflammation)和慢性炎症(chronic inflammation)。急性炎症起病急骤,持续时间短,仅几天到一个月,以渗出病变为其特征,炎症细胞浸润以粒细胞为主。慢性炎症持续时间较长,常数月到数年,常以增生病变为主,其炎症细胞浸润则以巨噬细胞和淋巴细胞为主。
鉴于抵抗病原微生物的两种主要成分,即白细胞和抗体均靠血液运输,因而在急性炎症中血液动力学改变、血管通透性增高和白细胞渗出这三种改变十分明显。结果造成富含蛋白质的渗出液、纤维蛋白及白细胞在损伤部位的血管外间隙积聚。这就是急性炎症病理组织学的主要特征。
一、血液动力学改变
组织受损伤后的微循环很快发生血液动力学变化,即血液的血管口径的改变,病变发展速度取决于损伤的严重程度。血液动力学的变化一般按下列顺序发生(图5-1)。
1.细去脉短暂收缩损伤发生后迅即发生短暂的细动脉收缩,持续仅几秒钟。其机制可能是神经源性的,但某些化学介质也能引起血管收缩。
2.血管扩张、血流加速先累及细动脉,随后导致更多微血管床开放,局部血流量增加,此乃急性炎症早期血液动力学改变的标志,也是局部红、热的原因。
血管扩张的发生机制与神经和体液因素均有关。神经因素即所谓轴突反射。以炎症介质所代表的体液因素对血管扩张的发生起更为重要的作用。
3.血流速度减慢血流速度减慢是微血管通透性升高的结果。富含蛋白质的液体向血管外渗出导致血管内红细胞浓集和粘稠度增加。最后扩张的小血管内挤满了红细胞,称为血流停滞(stasis)。
4.白细胞附壁随血流停滞的出现,微血管血液中的白细胞,主要是中性粒细胞始边集(leukoctytic margination)并与内皮细胞粘附,这一现象称为白细胞附壁。随后白细胞借阿米巴样运动游出血管进入组织间隙。
血液动力学变化所经历的时间与刺激的种类和强度有关。极轻度刺激所引起的血流加快仅持续10~15分钟,然后逐渐恢复正常;轻度刺激下血流加快,可持续几小时,接着血流变慢,甚至停滞;较重刺激下可在15~30分钟出现血流停滞,而严重损伤常仅需几分钟就可出现血流停滞。此外,局部血液动力学改变还与距离损伤因子远近有关,例如皮肤烧伤病灶的中心可能已发生了血流停滞,而周边部的血管尚处于扩张状态。
![]()
血管扩张,血流加快

血管进一步扩张,血流开始变慢,血浆渗出

血流变慢,白细胞游出血管外
血流显著变慢,除白细胞游出外,红细胞漏出
图5-1 血液动力学变化模式图
二、血管通透性升高
炎性水肿除了在炎症的最早阶段是由于血管扩张、血流速度加快导致流体静力压升高、血浆超滤,使基本不含蛋白质的液体从毛细血管滤出所致外,富含蛋白质的渗出液的产生则主要是由血管通透性增加造成的。由于大量蛋白质从血浆到达血管外间质,使血浆胶体渗透压降低,而组织胶体渗透压升高,使更大量液体聚集在间质内,从而形成炎性水肿或浆膜腔炎性积液。
微循环血管通透性的维护,主要依赖于内皮细胞的完整性。在炎症过程中,下列机制可引起血管通透性的增加(图5-2)。
1.内皮细胞收缩在组胺、缓激肽和其它炎症介质与内皮细胞受体结合后,可迅速引起内皮细胞收缩,致使内皮细胞间形成宽约0.5~1.0μm的缝隙。由于这些炎症介质的半寿期较短仅15~30分钟,故这种反应被称为速发短暂反应(immediate transient response)。此反应仅累及20~60μm口径的细静脉,而细动脉和毛细血管不受累。抗组胺药物能抑制此反应。
2.直接内皮损伤如严重烧伤和化脓菌感染等严重刺激可直接造成内皮细胞损伤,使之坏死和脱落。血管通透性增加发生迅速,并在高水平上持续几小时到几天,直至受损血管内形成血栓,此过程被称作速发持续反应(immediate-sustained response)。细动脉、毛细血管和细静脉各级微循环血管均可受累。
轻、中度热损伤、X线和紫外线损伤以及某些细菌毒素所引起的内皮细胞直接损伤等则发生较晚,常在2~12小时之后,但可持续几小时到几天,称为迟发持续反应(delayed prolonged response)。此反应仅累及毛细血管和小静脉。
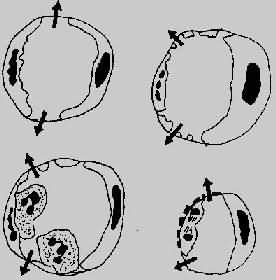
图5-2 血管通透性增加的四种机制模式图
左上图示内皮细胞收缩,累及细静脉;
右上图示直接损伤内皮细胞,累及全部微循环;
左下图示白细胞介导之内皮损伤,主要累及细静脉和毛细血管;
右下图示再生内皮细胞,主要累及毛细血管
3.白细胞介导的内皮损伤在炎症的早期,白细胞附壁并与内皮细胞粘附,引起白细胞的激活,释放具有活性的氧代谢产物和蛋白水解酶。后者可引起内皮细胞的损伤或脱落,使血管通透性增加。
4.新生毛细血管壁的高通透性在修复过程中所形成的新生毛细血管芽,其内皮细胞连接发育不成熟,可说明愈复性炎症中的液体外渗和水肿。
应该指出,上述四种机制在对某些刺激的反应过程中均发挥其作用。例如在热损伤的不同阶段,血管通透性增加所导致的液体外渗,可由化学介质造成的内皮细胞收缩、白细胞介导的内皮损伤、内皮的直接损伤和新生毛细血管壁的通透性引起。不同化介质可能相继被激活,从而导致持续反应。
局部炎性水肿可稀释毒素,减轻对局部的损伤作用;为局部浸润的白细胞带来葡萄糖、氧等营养物质,并带走代谢产物;渗出物所含的抗体和补体等物质,有利于消灭病原体;渗出物中的纤维蛋白原所形成的纤维素交织成网,一方面可限制病原微生物的扩散,使病灶局限化,另一方面也有利于吞噬细胞发挥吞噬作用,在炎症的后期纤维素网还可成为修复的支架,并有利于纤维母细胞产生胶原纤维;渗出物中的病原微生物和毒素随淋巴液被携带到局部淋巴结,可刺激机体产生细胞和体液免疫。
但如果渗出液过多,则可影响器官功能和压迫邻近器官,例如肺胞内渗出物堆集可影响换气功能,心包和胸腔积液可压迫心、肺、严重的喉头水肿可引起窒息,等等。纤维素性渗出物如果不能被完全吸收,则发生机化,例如在肺可引起肺肉质变在浆膜腔可引起浆膜粘连甚至浆膜腔闭锁。
三、白细胞的渗出和吞噬作用
炎症反应的最重要功能是将炎症细胞输送到炎症局部,白细胞的渗出是炎症反应最重要的特征。中性粒细胞和单核细胞渗出可吞噬和降解细菌、免疫复合物和坏死组织碎片,构成炎症反应的主要防御环节。但白细胞也可通过释放酶、化学介质和毒性自由基等,引起组织损伤并可能延长炎症过程。
白细胞的渗出过程是极其复杂的,经过附壁、粘着、游出和趋化作用等阶段到达炎症灶,在局部发挥重要的防御作用。
1.附壁随着血管扩张、血管通透性增加和血流缓慢,白细胞离开轴流,并沿内皮滚动。此时内皮细胞表面衬覆一层滚动的白细胞,犹如在人行道上前进的人群。最后白细胞粘附于血管内皮细胞。
2.粘着虽然多种因素影响着内皮细胞与白细胞的粘着,诸如内皮细胞和白细胞表面负电荷被中和而相互排斥力下降,二价阳离子桥接内皮细胞与白细胞而促进粘着等,但现在明了这种粘着是内皮细胞和白细胞表面粘附分子(adhesion motecule)相互识别引起的。炎平可使内皮细胞和炎症细胞表达新的粘附分子,增加粘附分子的数目和增强彼此的亲合性。
某些因子作用于内皮细胞,而另一些作用于白细胞,还有一些作用于两者,促进粘附分子的表达。
(1)白细胞表面粘附分子的表达:在补体C5a作用下白细胞增加三种整合蛋白类糖蛋白的表达。整合蛋白是由不同的α单位和β亚单位构成的异二聚体,具有广泛的生物功能。促进白细胞与内皮细胞粘着而表达于白细胞的整合蛋白包括LFA-1、MAC-1和P150-95(即CD11/CD18复合物)。C5a不仅可促进这三种整合蛋白的表达,还可改变其构象而增加与配体的亲合性。
(2)内皮细胞表面粘附分子的表达:在内皮细胞表面MAC-1和LFA-1的配体是细胞间粘附分子1(intercellular adhesion molecule 1,ICAM-1)。在IL-1和其它一些炎症介质的作用下,内皮细胞可增加细胞表面粘附分子的表达。高表达内皮细胞白细胞粘附分子1(endothelial leukocyte adhesion molecule 1,ELAM-1)可促进中性粒细胞的粘着;高表达ICAM-1促进中性粒细胞和淋巴细胞的粘着;血管细胞粘附分子(vascular cell adhesion molecule1,VCAM-1)促进淋巴细胞和单核细胞粘着。
(3)肿瘤坏死因子(TNF)则可促进内皮细胞和白细胞粘附分子的表达。
3.游出和趋化作用白细胞通过血管壁进入周围组织的过程称为游出(emigration)。粘着于内皮细胞表面的白细胞沿内皮表面缓慢移动,在内皮细胞连接处伸出伪足,整个白细胞逐渐以阿米巴运动方式从内皮细胞缝隙逸出,到达内皮细胞和基底膜之间,最终穿过基底膜到血管外(图5-3)。用电子显微镜可追踪此游出轨迹。一个白细胞通常需2~12分钟才能完全通过血管壁。中性粒细胞、单核细胞、淋巴细胞、嗜酸性和嗜碱性粒细胞都是以此种阿米巴运动方式游出的。血管壁受严重损伤时红细胞也可漏出,但这是个被运过程,是流体静压力把红细胞沿白细胞游出的途径或内皮细胞坏死崩解的裂口推出血管外。

图5-3 中性粒细胞的附壁和游出
注入致炎因子(毒素)10小时后的局部小静脉,可见四个中性粒细胞,1.中性粒细胞与多个红细胞(RBC)位于血管腔内,2.中性粒细胞正穿过血管壁到达血管外,3.中性粒细胞位于内皮细胞(E)和周细胞(P)之间,4.中性粒细胞游出达到血管外
炎症的不同阶段,游出的白细胞也不同。在急性炎症的早期,中性粒细胞首先游出。48小时后组织内则以单核细胞浸润为主,其原因首先在于中性粒细胞的寿命短,经24~48小时中性粒细胞崩解消失,而单核细胞在组织内存活时间长;中性粒细胞停止游出后,单核细胞仍可持续游出;第三个因素为在炎症的不同阶段所激活的趋化因子不同。现已证实中性粒细胞能释放单核细胞趋化因子,因此中性粒细胞游出后必然引起单核细胞的游出。此外,由于致炎因子不同,渗出的白细胞也不同:常见的葡萄球菌和链球菌感染,以中性粒细胞渗出为主;病毒感染以淋巴细胞为主;在一些过敏反应,则以嗜酸性粒细胞渗出为主。
趋化作用(chemotaxis)是指白细胞向着化学刺激物所在部位作定向移动,移动速度约每分钟5~20μm。这些化学刺激物称为趋化因子。研究白细胞的趋化作用可采用微孔滤膜Boyden小室技术的和显微缩时摄影技术记录白细胞的运动轨迹。研究发现,趋化因子的作用是有特异性的,有些趋化因子只吸引中性粒细胞,而另一些趋化因子则吸引单核细胞,或嗜酸性粒细胞等。此外,不同细胞对趋化因子的反应能力也不同。粒细胞和单核细胞对趋化因子的反应较显著。而淋巴细胞对趋化因子的反应则较弱。
一些外源性和内源性化学物质具有趋化作用。常见的白细胞趋化因子包括可溶性细菌产物、补体系统成分(特别是C5a)和花生四烯酸经脂质加氧酶途径的代谢产物(特别是白细胞三烯B4)等。单核细胞还对中性粒细胞的衍生物、致敏淋巴细胞所释放的因子及纤维粘连蛋白断片起趋化反应。在Ⅰ型变态反应中,IgE致敏的肥大细胞或嗜碱性粒细胞在同种抗原的刺激下,可释放过敏反应嗜酸性粒细胞趋化因子(EFC-A),致使嗜酸性粒细胞积聚。此外致敏淋巴细胞释放的因子和C5a也是嗜酸性粒细胞的趋化因子。
白细胞如何“发现”趋化因子,趋化因子又如何引起白细胞定向运动?首先,白细胞表面有趋化因子的受体,在各种趋化因子与其特异性受体结合后可引起一系列信号传导通路成分的改变,尽管其全部机制并未彻底揭开。在趋化因子与白细胞表面的受体激活后可激活磷脂酶C,导致4,5二磷酸磷脂酰肌醇水解,产生三磷酸肌醇和二乙酰基甘油,进而使细胞内游离钙离子升高,首先是细胞内储存钙释放,然后是细胞外钙离子通过钙通道进入细胞内。由于细胞内钙离子浓度升高,细胞内便组装可引起细胞运动的收缩成分。此外在趋化因子与受体结合后还能激活磷脂酶A2,后者将细胞膜磷脂转换成花生四烯酸。二乙酰基甘油通过激活蛋白激酶C在白细胞激活的不同阶段、脱颗粒和分泌过程中发挥作用。
4.白细胞在局部的作用游出的白细胞在炎症灶局部发挥吞噬作用(phagocytosis)和免疫作用,能有效地杀伤病原微生物,因而成为炎症防御反应中极其重要的一环。
(1)吞噬作用:吞噬作用是指白细胞游出到炎症灶,吞筮病原体以及组织碎片的过程。完成此功能的吞噬细胞主要有两种;中性粒细胞和巨噬细胞,其吞噬异物的过程基本相同,但其结构和化学成分则有所不同。
1)吞噬细胞的种类:中性粒细胞直径10~12μm,胞核呈杆状或分叶状,通常为2~5叶,叶间有染色质丝相连,核染色质呈块状,着色深。细胞浆内富含中性颗粒,相当于电镜下的溶酶体。在电镜下,中性颗粒可分为嗜苯胺蓝颗粒和特异性颗粒两种。前者又称嗜天青颗粒,体积较大、电子密度高,约占全部颗粒的10%~20%。含有酸性水解酶、中性蛋白酶、髓过氧化物酶、阳离子蛋白、溶菌酶及磷脂酶A2等。特异性颗粒较小、电子密度较低,占全部颗粒的80%,含有溶菌酶、磷脂酶A2、乳铁蛋白及碱性磷酸酶等。
炎症灶内的巨噬细胞,大多来自血液的单核细胞,直径为14~17μm。细胞核呈肾形或弯曲折叠的不规则形,染色质颗粒纤细而疏松,故着色较浅。胞浆较丰富,内有大小、致密度和形态均不一致的溶酶体,富含酸性磷酸酶和氧化酶。巨噬细胞受外界刺激能被激活,表现为细胞体积增大,细胞表面皱襞增多,线粒体和溶酶体增多,其功能也相应增强。
2)吞噬过程:包括识别和粘着、吞入及降解三个阶段。
①识别和粘着:在无血清存在的条件下,吞噬细胞很难识别并吞噬细菌。因为在血清中存在着调理素(opsonin),即一类能增强吞噬细胞吞噬活性的血清蛋白质,主要是IgG和C3b。吞噬细胞藉其表面的Fc受体和C3b(C3bi或Mac-1),能识别被抗体或补体包被的细菌,经抗体或补体与相应受体结合,细菌就被粘着在吞噬细胞的表面(图5-4)。
②吞入:细菌粘着于吞噬细胞表面之后,吞噬细胞乃伸出伪足,随伪足延伸和互相吻合,形成由吞噬细胞包围吞噬物的泡状小体,谓之吞噬体(phagosome)。吞噬体逐渐脱离细胞膜进入细胞内部,并与初级溶酶体融合,形成吞噬溶酶体(phagolysosome),溶酶体内容物倾注其中,细菌在吞噬溶酶体内被杀伤、降解。
③杀伤和降解:进入吞噬溶酶体的细菌主要是被具有活性的氧化代谢产物杀伤的。吞噬过程使白细胞的耗氧量激增,可达正常消耗量的2~20倍,并激活白细胞氧化酶(NADPH氧化酶),后者使还原型辅酶Ⅱ(NADPH)氧化而产生超氧负离子(O2-)。
![]()
大多数超氧负离子经自发性歧化作用转变为H2O2。在中性粒细胞的嗜天青颗粒中存在着髓过氧化物酶(MPO),在有氯化物存在的条件下,该酶可将H2O2还原生成次氯酸HOCL·)(图5-5)。
![]()
HOCL·是强氧化剂和杀菌因子。羟自由基是另一种杀菌因子。
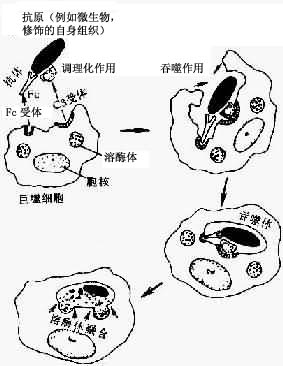
图5-4吞噬过程示意图
示吞噬细胞的识别和吞入
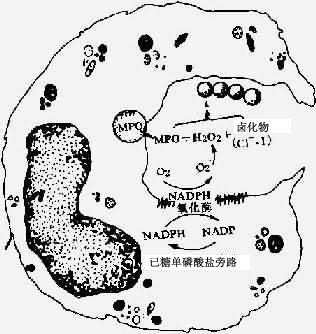
图5-5 吞噬细胞的氧化代谢活性产物杀菌机制图解
氧代谢产物可通过下列途径杀菌:
①与细菌细胞膜磷脂分子中的高度不饱和脂肪酸发生脂质过氧化反应,导致细胞膜的生理状态破坏,细胞膜对阳离子的通透性升高。细菌内游离钙离子浓度的升高,可激活钙依赖性磷脂酶和某些蛋白激酶,从而造成细菌的损伤和死亡。
②与氨基酸、蛋白质和糖分子上的某些反应基因发生氧化还原反应,使具有重要生理功能的酶失去活性,一些大分子物质改变其物理和生物学特性。
③可穿过细胞膜进入细胞内部,与细菌内部分子起作用。
④与细菌的DNA发生反应,促进姐妹染色单体交换。此外脂质过氧化物在分解过程中产生丙二醛,后者与DNA发生交联,从而影响DNA复制,阻断细菌繁殖,最后导致细菌死亡。
因此,H2O2-MPO-CL-系统是最有效的杀菌系统,其杀菌效能比单独H2O2强50倍,而且对细菌、真菌、支原体和病毒均有杀伤效应。
白细胞颗粒中那些不依赖氧的物质也能杀伤病原体,包括增加细菌通透性蛋白(bacterial permeability-increasing protein,即BPI蛋白)、溶菌酶(水解细胞之细胞壁)、乳铁蛋白和一组新发现的富含精氨酸的阳离子蛋白质,后者能溶解细菌细胞壁,被称作杀菌(phagocytin)或防御素(defensins)。吞噬作用完成后,吞噬溶酶体内的pH降至4~5,其内的酸性水解酶就可在此种合适的的pH环境下发挥降解细菌的作用。
通过吞噬细胞的上述杀伤作用,大多数病原微生物被杀伤。但有些细菌,在白细胞内处于静止状态,仍具有生命力和繁殖力,如结核杆菌,一旦机体抗抵力下降,这些病原体又能繁殖,并可随吞噬细胞的游走而在体内播散。生活在吞噬细胞内的细菌难以受到抗生素和机体防御机制的影响,故很难在机体内消灭。
(2)免疫反应:免疫反应需淋巴细胞、浆细胞和巨噬细胞的协同作用。淋巴细胞大小不一,直径6~16μm。胞核圆形或卵圆形,核的一侧常有小凹陷,核染色质呈致密块状,故着色深。胞浆少,可见少数不含过氧化酶的嗜苯胺蓝颗粒。淋巴细胞又分为T细胞和B细胞。浆细胞形状特殊,呈一端稍粗的卵圆形,核呈圆形,位于细胞的较粗端一侧,在胞浆侧与胞浆间有一亮晕核的异染色质丰富呈轮辐状排列。胞浆略嗜碱性,电镜下富含粗面内质网。其功能在于产生抗体。
抗原进入机体后,巨噬细胞将其吞噬处理,再把抗原递呈给T和B细胞,使其致敏。免疫活化的淋巴细胞分别产生淋巴因子和抗体,发挥其杀伤病原微生物的作用。
淋巴细胞和浆细胞浸润常见于慢性炎症,尤其是与细胞免疫有关的慢性肉芽肿性炎症,如结核、梅毒等。
(3)组织损伤:在某些情况下,白细胞激活后可向细胞外间隙释放其产物,这些产物包括溶酶体酶、氧源性代谢产物和花生四烯酸代谢产物(前列腺素和白细胞三烯)等。这些产物本身有强烈的介导内皮细胞和组织损伤的作用及加重原始炎症刺激因子的损伤效能。这种白细胞介导的组织损伤在许多人类炎症性疾病中都能见到,类风湿性关节炎便是一个明显的例证。
5.白细胞的功能缺陷如前所述,白细胞在机体的防御反应中起着极其重要的作用。先天性和获得性白细胞功能缺陷将造成病人的反复感染。从白细胞先天性功能缺陷的病例可加深对白细胞在炎症反应中的作用的认识。现举例说明如下:
(1)吞噬功能缺陷:可表现在从与内皮细胞的粘着到杀菌活性的全过程。
1)粘着缺陷:典型的例子是白细胞粘着缺陷(leukocyte adhesion deficiency), 见于常染色体隐生遗传病,其发病机制在于LFA-1和Mac-1中的β链生物合成缺陷,因而影响白细胞粘着于内皮细胞。
2)识别障碍:主要因调理素缺乏,见于丙种球蛋白缺乏症及补体缺乏。
3)趋化作用缺陷:可表现为白细胞运动能力降低和趋化因子产生障碍,如在Chédiak-Higashi综合征(常染色体隐性遗传性疾病),以白细胞胞浆内出现巨大溶酶体为其特征,存在多种异常,包括微管组装障碍,因而影响白细胞的位移。而先天性补体缺乏则使趋化因子生成障碍。
4)吞入或脱颗粒障碍:在Chédiak-Higashi综合征时,由于微管组装障碍,而影响溶酶体内容物倾入吞噬小体,影响白细胞的杀菌能力,因而病人表现为反复化脓性感染。中性粒细胞肌动蛋白功能障碍(actin dysfunction)影响病原体的吞入。当然,调理素缺乏亦影响病原体的吞入。
5)杀伤作用缺陷:H2O2产生障碍见于慢性肉芽肿病(chronic granulomatous disease,CGD),它是X连锁遗传性疾病,男性婴儿和儿童患病,是由于NADPH氧化酶缺乏,故影响H2O2的产生,导致H2O2-MPO-CL-杀菌功能不良。一般细菌在代谢过程中能产生少量H2O2,但有的细菌能产生分解H2O2的过氧化氢酶,如金黄色葡萄球菌,所以该细菌可在CGD患者体内存活下来。而肺炎双球菌等产生少量H2O2,却不产生过氧化氢酶,这些H2O2则足以启动H2O2-MPO-CL-杀菌功能系统,而将细菌杀死。因此CGD病人常表现为过氧化氢酶阳性细菌的感染。这类病例说明活性氧化代谢产物在杀灭病原微生物中的重要作用。
某些疾病如Chédiak-Higashi综合征和糖尿病,则因多个环节缺陷而影响吞噬作用。
(2)免疫反应缺陷:主要是先天性免疫缺陷,严重损伤机体的免疫功能及炎症反应。例如B细胞缺陷(Bruton综合征)、T细胞缺陷(Di George综合征),以及联免疫缺陷病等。
了解急性炎症过程的基本病变就可清楚地阐明炎症的局部表现。炎症局部小血管呈明显而又持续性地扩张,致使局部组织发红。发生在体表或接近皮肤的炎症,由于局部血流量增多,故局部发热。肿胀的主要原因是局部水肿和渗出物积聚。对局部疼痛的机制目前还不十分清楚,某些炎症的介质如缓激肽和某些前列腺素可引起疼痛。因局部水肿和渗出物积聚所引起的组织内张力增高可能是疼痛最重要的因素,因此脓肿在局部排脓后疼痛症状可立即缓解。局部功能障碍是由于疼痛而反射性地抑制肌肉活动,以及局部水肿使活动受限所致。
四、炎症介质
急性炎症反应中的血管扩张、通透性升高和白细胞渗出的发生机制,是炎症发生机制的重要课题。有些致炎因子可直接损伤内皮,引起血管通透性升高,但许多致炎因子并不直接作用于局部组织,而主要是通过内源性化学因子的作用而导致炎症,故又称之为化学介质或炎症介质(inflammatory mediator)。
1.细胞释放的炎症介质
(1)血管活性胺:包括组胺和5-羟色胺(5-HT)。组胺主要存在于肥大细胞和嗜碱性粒细胞的颗粒中,也存在于血小板。引起肥大细胞释放组胺的刺激包括:①创伤或热等物理因子;②免疫反应,即抗原与结合于肥大细胞表面的IgE相互作用时,可使肥大细胞释放颗粒;③补体片断,如过敏毒素(anaphylatoxin);④中性粒细胞溶酶体阳离子蛋白;⑤某些神经肽。在人类,组胺可使细动脉扩张,细静脉内皮细胞收缩,导致血管通透性升高。组胺可被组胺酶灭活。组胺还有对嗜酸性粒细胞的趋化作用。
5-HT由血小板释放,胶原和抗原抗体复合物可刺激血小板发生释放反应。虽然在大鼠其作用与组胺相似,但在人类炎症中的作用尚不十分清楚。
(2)花生四烯酸代谢产物:包括前列腺素(PG)和白细胞三烯(leukotriene,LT),均为花生四烯酸(arachidonic acid,AA)的代谢产物。AA是二十碳不饱和脂肪酸,是在炎症刺激和炎症介质(如C5a)的作用下激活磷脂酶产生的,在炎症中,中性粒细胞的溶酶体是磷脂酶的重要来源。AA经环加氧酶和脂质加氧酶途径代谢,生成各种产物(图5-6)。
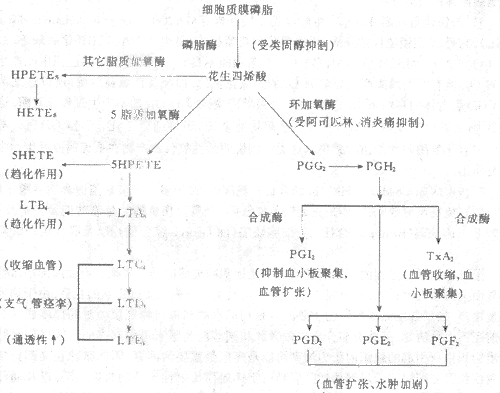
图5-6 炎症过程中花生四烯酸的代谢
总之,炎症刺激花生四烯酸代谢并释放其代谢产物,导致发热、疼痛、血管扩张、通透性升高及白细胞渗出等炎症反应。另一方面,抗炎药物如阿司匹林、消炎痛和炎固醇激素则能抑制花生四烯酸代谢、减轻炎症反应。
(3)白细胞产物 被致炎因子激活后,中性粒细胞和单核细胞可释放氧自由基和溶酶体酶,促进炎症反应和破坏组织,成为炎症介质。
1)活性氧代谢产物:其作用包括三个方面:①损伤血管内皮细胞导致血管通透性增加。②灭活抗蛋白酶(如可灭活α1抗胰蛋白酶),导致蛋白酶活性增加,可破坏组织结构成分,如弹力纤维。③损伤红细胞或其它实质细胞。
当然,血清、组织液和靶细胞亦有抗氧化保护机制,故是否引起损伤取决于两者之间的平衡状态。
2)中性粒细胞溶酶体成分:因中性粒细胞的死亡、吞噬泡形成过程中的外溢及出胞作用,溶酶体成分可外释,介导急性炎症。其中中性粒细胞蛋白酶,如弹力蛋白酶、胶原酶和组织蛋白酶可介导组织损伤。
阳离子蛋白质具有如下生物活性:①引起肥大细胞脱颗粒而增加血管通透性;②对单核细胞的趋化作用;③起中性和嗜酸性粒细胞游走抑制因子的作用。
(4)细胞因子:细胞因子(cytokines)主要由激活的淋巴细胞和单核细胞产生,可调节其他类型细胞的功能,在细胞免疫反应中起重要作用,在介导炎症反应中亦有重要功能。
IL-1和TNF的分泌可被内毒素、免疫复合物、物理性损伤等多种致炎因子刺激,可通过自分泌、旁分泌和全身作用等方式起作用。特别是它们可促进内皮细胞表达粘附分子,增进白细胞与之粘着。也可以引起急性炎症的发热。TNF还能促进中性粒细胞的聚集和激活间质组织释放蛋白水解酶。IL-8是强有力的中性粒细胞的趋化因子和激活因子(图5-7)。
图5-7 IL-1和TNF在炎症中的主要作用
(5)血小板激活因子:血小板激活因子(platelet activating factor,PAF)是另一种磷脂起源的炎症介质,乃由IgE致敏的嗜碱性粒细胞在结合抗原后产生。除了能激活血小板外,PAF可增加血管的通透性、促进白细胞聚焦和粘着,以及趋化作用。此外还具有影响全身血液动力学的功能。嗜碱性粒细胞、中性粒细胞、单核细胞和内皮细胞均能释放PAF。PAF一方面可直接作用于靶细胞,还可刺激细胞合成其他炎症介质,特别是PG和白细胞三烯的合成。
(6)其他炎症介质:P物质可直接和间接刺激肥大胞脱颗粒而引起血管扩张和通透性增加。内皮细胞、巨噬细胞和其他细胞所产生的一氧化氮可引起血管扩张和具细胞毒性。
2.体液中产生的炎症介质血浆中有三种相互关联的系统,即激肽、补体和凝血系统;为重要的炎症介质。
(1)激肽系统:激肽系统的激活最终产生缓激肽(bradykinin),后者可引起细动脉扩张、内皮细胞收缩、细静脉通透性增加,以及血管以外的平滑肌收缩。缓激肽很快被血浆和组织内的激肽酶灭活,其作用主要局限在血管通透性增加的早期。
(2)补体系统;补体系统由一系列蛋白质组成,补体的激活有两种途径—经典和替代途径。在急性炎症的复杂环境中,下列因素可激活补体:①病原微生物的抗原成分与抗体结合通过经典途径激活补体,而革兰氏阴性细菌的内毒素则通过替代途径激活补体。此外,某些细菌所产生的酶也能激活C3和C5。②坏死组织释放的酶能激活C3和C5。③激肽、纤维蛋白形成和降解系统的激活及其产物也能激活补体。
补体可从以下三个方面影响急性炎症:①C3a和C5a(又称过敏毒素)增加血管的通透性,引起血管扩张,都是通过引起肥大细胞和单核细胞进一步释放炎症介质;C5a还能激活花生四烯酸代谢的脂质加氧酶途径,使中性粒细胞和单核细胞进一步释放炎症介质;②C5a引起中性粒细胞粘着于血管内皮细胞,并且是中性粒细胞和单核细胞的趋化因子;③C3b结合于细菌细胞壁时具有调理素作用,可缯强中性粒细胞和单核细胞的吞噬活性,因为在这些吞噬细胞表面有C3b的受体。
C3和C5是最重要的炎症介质。除了前述的激活途径外,C3和C5还能被存在于炎症渗出物中的蛋白水解酶激活,包括纤维蛋白溶酶和溶酶体酶。因此而形成中性粒细胞游出的不休止的环路,即补体对中性粒细胞有趋化作用,中性粒细胞释放的溶酶体又能激活补体。
(3)凝血系统:Ⅻ因子激活不仅能启动激肽系统,而且同时还能启动血液凝固和纤维蛋白溶解两个系统。凝血酶在使纤维蛋白原转化为纤维蛋白的过程中释放纤维蛋白多肽,后者可使血管通透性升高,又是白细胞的趋化因子。
图5-8 激肽、凝血、纤维蛋白溶解及补体的相互作用
纤维蛋白溶解系统可通过激肽系统引起炎症的血管变化。由内皮细胞、白细胞和其他组织产生的纤维蛋白溶酶原激活因子,能使纤维蛋白溶酶原转变成纤维蛋白溶酶,后者通过如下三种反应影响炎症的进程:①通激活第Ⅻ因子启动缓激肽的生成过程;②裂解C3产生C3片断;③降解纤维蛋白产生其裂解产物,进而使血管通透性增加。
炎症介质的作用有两点值得注意。第一,不同介质系统相互之间有着密切的联系,例如图5-8所示的补体、激肽及凝血系统和纤维蛋白溶解系统的激活和其产物的密切关系,便是例证。这些炎症介质的作用也是交织在一起的。第二,几乎所有介质均处于灵敏的调控和平衡体系中。一方面在细胞内处于严密隔离状态的介质,或在血浆和组织内处于前体状态的介质,都必须经过许多步骤才能被激活,在其转化过程中,限速机制控制着产生介质的生化反应的速度。另一方面,介质一旦被激活和被释放,将迅速被灭活或破坏。机体就是通过这种调控体系使体内介质处于动态平衡。
主要炎症介质的的作用小结于表5-1。
表5-1 主要炎症介质的作用
| 功 能 | 炎症介质种类 |
| 血管扩张 | 组胺、缓激肽、PGI2、PGE2、PGD2、PGF2α |
| 血管通透性升高 | 组胺、缓激肽、C3a、C5a白细胞三烯C4、D4、E4、PAF、活性氧代谢产物 |
| 趋化作用 | 白细胞三烯B4、C5a、细菌产物、中性粒细胞阳离子蛋白细胞因子(IL-8、TNF) |
| 发热 | 细胞因子(IL-1、TNF) |
| 疼痛 | PGE2、缓激肽 |
| 组织损伤 | 氧自由基、溶酶体酶 |
五、急性炎症的类型
由于致炎因子的不同、组织反应轻重程度的不同和炎症的发生部位不同,急性炎症的病理形态也不同。根据渗出物的主要成分,急性炎症分为浆液性炎、纤维素性炎、化脓性炎和出血性炎。
1.浆液性炎症浆液性炎(serous inflammation)以血清渗出为其特征,渗出的主要成分为浆液,其中混有少量白细胞和纤维素。浆液内含有3%~5%的蛋白质,主要是白蛋白。浆液性炎常发生于疏松结缔组织、浆膜和粘膜等处。浆液性渗出物弥漫地浸润于组织内,局部出现明显的炎性水肿,如毒蛇咬伤、皮肤二度烧伤时渗出液蓄积于表皮内,形成水疱。体腔的浆液性炎造成炎性积液,浆液不仅来自血管渗出,而且也来自间皮细胞的分泌增加,如结核性胸膜炎、风湿性关节炎等。粘膜的浆液性炎又称浆液性卡他,如见于感冒初期的鼻炎。卡他(catarrh)一词来自希腊语,是向下滴流的意思。一般用于粘膜的渗出性炎症,形容渗出液较多,沿粘膜表面向外排出。浆膜或粘膜浆液性炎时,间皮或上皮细胞可发生变性、坏死和脱落。
浆液性炎一般较轻,易于消退。但有时因浆液渗出过多可导致严重后果,如胸腔和心包腔内有大量浆液时,可影响呼吸和心功能。
2.纤维素性炎症纤维素性炎症(fibrinous inflammation)时以纤维蛋白原渗出并在炎症灶内形成纤维素为主。光镜下,苏木素伊红染色可见大量红染的纤维素交织呈网状,间隙中有中性粒细胞及坏死细胞的碎屑。大片纤维素在镜下表现为片状、红染、质地均匀的物质。纤维蛋白原的大量渗出,说明血管壁损伤较重,多由于某些细菌毒素(如白喉杆菌、痢疾杆菌和肺炎双球菌的毒素)或各种内源性或外源性毒物质(如尿毒症时的尿素和汞中毒)所引起。病变常发生于粘膜、浆膜和肺。在粘膜的纤维素性炎(如白喉、细菌性痢疾),纤维素、白细胞和坏死的粘膜上皮常混合在一起,形成灰白色的膜状物,称为假膜。因此,粘膜的纤维素性炎又称为假膜性炎(图5-9,图5-10)。由于局部组织结构的特点不同,有的假膜牢固附着于粘膜面不易脱落(如咽白喉),有的假膜却与粘膜损伤部联系松散,容易脱落(如气管白喉),脱落的假膜可堵塞支气管而引起窒息。浆膜的纤维素性炎常见于胸膜腔和心包腔,如肺炎双球菌引起的纤维素性胸膜炎及风湿性心包炎。在心包的纤维素性炎时,由于心脏的搏动,使心外膜上的纤维素形成无数绒毛状物,覆盖于心表面,因而又有“绒毛心”之称。此外,大叶性肺炎的红色和灰色肝样变期均有大量纤维蛋白原渗出。
图5-9 白喉 咽喉及气管和支气管粘膜表面有假膜覆盖

图5-10 纤维素性胸膜炎
胸膜表面覆盖大量纤维素性渗出物
少量的纤维素可以被中性粒细胞释放的溶蛋白酶溶解吸收。但是,正常血清和组织中含有一定量的抗胰蛋白酶,这就在一定程度上对抗中性粒细胞溶蛋白酶的作用。因此,如果纤维素较多,加之中性粒细胞所释出的溶蛋白酶较少或组织内抗胰蛋白酶较多时,纤维素不可能被完全溶解吸收,结果发生机化(organization),引起浆膜增厚和粘连,甚至浆膜腔闭锁,严重影响器官功能。
3.化脓性炎症化脓性炎症(suppurative or purulent inflammation)以中性粒细胞大量渗出,并伴有不同程度的组织坏死和脓液形成为特征。多由葡萄球菌、链球菌、脑膜炎双球菌、大肠杆菌等化脓菌引起,亦可因某些化学物质(如松节油)和机体坏死组织所致。临床上常见的化脓性炎症有疖、痈、化脓性阑尾炎和化脓性脑膜炎等。脓性渗出物称为脓液(pus),是一种混浊的凝乳状液体,呈灰黄色或黄绿色。由葡萄球菌引起的脓液,其质浓稠,而由链球菌引起的脓液,则较稀薄。脓液中的中性粒细胞除少数仍可保持其吞噬能力外,大多数已发生变性和坏死,即变为脓细胞。脓液中除脓细胞外,还含有细菌、被溶解的坏死组织碎屑和少量浆液。根据化脓性炎症发生的原因和部位的不同,可将其分为下列三类。
(1)表面化脓和积脓:表面化脓是指浆膜或粘膜组织的化脓性炎。粘膜化脓性炎又称脓性卡他。此时,中性粒细胞主要向粘膜表面渗出,深部组织没有明显的炎性细胞浸润,如化脓性尿道炎或化脓性支气管炎,渗出的脓液可通过尿道、气管而排出体外。当这种病变发生在浆膜或胆囊、输卵管的粘膜时,脓液则在浆膜腔或胆囊、输卵管腔内蓄积,称为积脓(empyema)。
(2)蜂窝织炎(phlegmonous inflammation):疏松组织中弥慢性化脓称为蜂窝织炎,常见于皮肤、肌肉和阑尾(图5-11)。蜂窝织炎主要由溶血性链球菌引起。链球菌能分泌透明质酸酶,降解结缔组织基质的透明质酸;分泌链激酶,溶解纤维素。因此,细菌易于通过组织间隙和淋巴管蔓延扩散造成弥漫性浸润。
(3)脓肿(abscess):为局限性化脓性炎症,主要特征为组织发生坏死溶解,形成充满脓液的腔,称为脓肿(图5-12,图5-13)。可发生在皮下或内脏,常由金黄色葡萄球菌引起。这些细菌能产生毒素使局部组织坏死,继而大量中性粒细胞浸润,以后粒细胞崩解释出酶将坏死组织液化,形成含有脓液的空腔。金黄色葡萄球菌还产生血浆凝固酶,能使渗出的纤维蛋白原转变为纤维素,因而病变比较局限。最近发现,金黄色葡萄球菌有层粘连蛋白受体,因而容易通过血管壁并引起转移性脓肿。小脓肿可以吸收消散,较大脓肿则由于脓液过多,吸收困难,需要切开排脓或穿刺抽脓,而后由肉芽组织修复,形成瘢痕。
疖是毛囊、皮脂腺及其附近组织所发生的脓肿。疖中心部分液化、变软后,脓肿就可以穿破。痈是多个疖的融集,在皮下脂肪、筋膜组织中形成的许多互相沟通的脓肿,必须及时切开引流排脓后,局部才能修复愈合。在皮肤或粘膜的化脓性炎时,由于皮肤或粘膜坏死、崩解脱落,可形成局部缺陷,即溃疡(ulcer)。深部脓肿如向体表或自然管道穿破,可形成窦道(sinus)或瘘管(fistula)。窦道是指只有一个开口的病理性盲管,瘘管是指连接于体外与有腔器官之间或两个有腔器官之间的、有两个以上开口的病理性管道。例如肛门周围组织的脓肿,可向皮肤穿破,形成脓性窦道,也可既向皮肤穿破,又向肛管穿破,形成脓性瘘管。脓性窦道或脓性瘘管不断排出脓性渗出物,长期不愈。
4.出血性炎症出血性炎(hemorrhagic inflammation)不是一种独立的炎症类型,只是当炎症灶内的血管壁损伤较重时,渗出物中才有大量红细胞,形成出血性炎症。常见于流行性出血热,钩端螺旋体病或鼠疫等。
上述各种类型的炎症可单独发生,在有些炎症过程中两种不同类型可以并存,如浆液纤维素性炎或纤维素性化脓性炎等。此外,在炎症发展过程中,一种类型炎症可转变为另一种类型,如从浆液性炎开始,可进一步发展成为纤维素性或化脓性炎。

图5-11 蜂窝织炎性阑尾炎
脓性渗出物弥漫浸润于阑尾的粘膜、粘膜下层、肌层及浆膜层
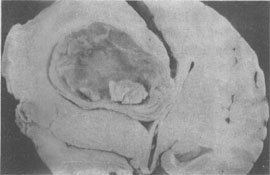
图5-12 脑脓肿
脑实质中有一大脓腔,腔内充满凝乳状脓汁
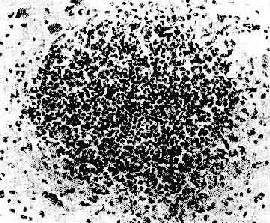
图5-13 脑脓肿
脓肿内脑组织完全坏死、液化,并有大量脓球聚焦
六、急性炎症的结局
在炎症过程中,如渗出和增生等抗损伤过程占优势,则炎症逐渐向痊愈方向发展;相反,如损伤性变化占优势,则炎症逐渐加重并可向全身扩散;若损伤和抗损伤变化暂时难分“胜负”,则炎症转变为慢性。
(一)痊愈
大多数炎症病变能够痊愈。
1.完全痊愈在炎症过程中,清除病因,溶解吸收少量的坏死物和渗出物,通过周围健康细胞的再生达到修复,最后完全恢复组织原来的结构和功能。
2.不完全痊愈如炎症灶的坏死范围较广,则由肉芽组织修复,留下瘢痕,不能完全恢复组织原有的结构和功能。
(二)迁延不愈,转为慢性
致炎因子不能在短期内清除、或在机体内持续存在,而且还不断损伤组织,造成炎症过程迁延不愈,急性炎症转化为慢性炎症,病情时轻时重。
(三)蔓延扩散
在病人的抵抗力低下,或病原微生物毒力强、数量多的情况下,病原微生物可不断繁殖并直接沿组织间隙向周围组织、器官蔓延,或向全身扩散。
1.局部蔓延炎症局部的病原微生物可经组织间隙或器官的自然通道向周围组织和器官扩散,如肾结核可沿泌尿道下行播散,引起输尿管和膀胱结核。
2.淋巴道扩散急性炎症时,从血管渗出的含蛋白液体可通过淋巴液回流入血,藉此可减轻或延缓水肿的发生。但在严重损伤的情况下,病原微生物可随淋巴液扩散,引起继发性淋巴管炎及所属淋巴结炎,例如足部感染时,下肢因淋巴管炎可出现红线,腹股沟淋巴结炎表现为局部肿大,并引起疼痛。淋巴道的这些变化有时可限制感染的扩散,但感染严重时,病原体可通过淋巴入血,引起血道扩散。
3.血道扩散炎症灶的病原微生物或某些毒性产物可侵入血循环或被吸收入血,引起菌血症、毒血症、败血症和脓毒性败血症等。
(1)菌血症(bacteremia):细菌由局部病灶入血,但全身并无中毒症状,从血液中可查到细菌,称为菌血症。一些炎症性疾病的早期都有菌血症,如大叶性肺炎等。在菌血症阶段,肝、脾、骨髓的吞噬细胞可组成一道防线,以清除病原体。
(2)毒血症(toxemia):细菌的毒素或毒性产物被吸收入血,为毒血症。临床上出现高热、寒战等中毒症状,同时伴有心、肝、肾等实质细胞的变性或坏死。严重时甚至出现中毒性休克。
(3)败血症(septicemia):毒力强的细菌进入血中不仅未被清除而且还大量繁殖,并产生毒素,引起全身中毒症状和病理变化,称为败血症。患者除有严重的毒血症临床表现外,还常出现皮肤、粘膜的多发性出血班点和脾及全身淋巴结肿大等。此时血液中常可培养出致病菌。
(4)脓毒败血症(pyemia):化脓菌可引起的败血症可进一步发展为脓毒败血症。此时除有败血症的表现外,同时还在一些器官(如肺、肾、肝等)形成多个脓肿。这些脓肿通常较小、较均匀散布在器官中。镜下,脓肿的中央及尚存的毛细血管或小血管中常见到细菌菌落,说明脓肿是由栓塞於器官毛细血管的化脓菌所引起,故称之为栓塞性脓肿(embolicabscess)或转移性脓肿(metastatic abscess)。
第三节 慢性炎症
致炎因子持续存在并且损伤组织是发生慢性炎症的根本原因。各种器官的慢性炎症除从急性炎症转化而来外,还可以其他方式发生。急性炎症反复发作,而发作间期无明显症状也是慢性炎症,如慢性胆囊炎、慢性肾盂肾炎等。慢性炎症还可潜隐缓慢地逐渐发生,临床上开始并无急性炎症表现,常见于细胞内感染(如结核杆菌、病毒感染),这些病原体的毒力不强,但可引起免疫反应;或长期受不能降解却有潜在毒性物质的刺激(如硅肺);或持续存在的、对抗自身组织的免疫反应即自身免疫性疾病(如类风湿性关节炎)。
一、一般慢性炎症的基本病理变化
1.炎症灶内主要是巨噬细胞、淋巴细胞和浆细胞浸润。单核吞噬细胞的浸润对慢性炎症十分重要。单核细胞从血管游出后转化为巨噬细胞。巨噬细胞还可被激活。在炎症灶局部巨噬细胞的积聚有三方面的原因:①由于从炎症灶不断产生吸引单核细胞的趋化因子,如C5a、纤维蛋白多肽、阳离子蛋白质及胶原和纤维粘连蛋白的分解产物等。因此,从血液循环中渗出的单核细胞源源不断来到局部,这是局部巨噬细胞的主要来源。②游出的巨噬细胞在局部通过有丝分裂而增殖,但巨噬细胞局部增殖的起始动因还不清楚。③炎症灶内的巨噬细胞寿命长,并能长期停留局部而不游走。单核细胞在细胞因子(如γ干扰素)、细菌内毒素和接触包被有纤维粘连蛋白界面等因子的作用下,可被激活。激活的单核细胞分泌多种生物活性产物,是造成慢性炎症中的组织破坏和纤维化的重要介质。
至于淋巴细胞和浆细胞在与免疫有关的炎症反应中的作用已很明确,但其在非免疫性炎症中的功能尚有待于研究。应当指出,中性粒细胞通常是急性炎症的标志,但在一些慢性炎症,也可见到大量中性粒细胞浸润,并可形成脓液;反之,淋巴细胞浸润也并非总是慢性炎症的特征,在急性病毒感染如急性病毒性肝炎时,淋巴细胞则为炎症浸润的主要成分。
单核细胞所产生的细胞因子可激活淋巴细胞,而激活的淋巴细胞可以产生炎症介质,也是造成慢性炎症持续的重要因素。
嗜酸性粒细胞在IgE介导的免疫反应和寄生虫感染时十分常见,其颗粒中所含的主要碱性蛋白(major basic protein,MBP)对寄生虫有毒性,而且可引起哺乳类动物的上皮细胞溶解。
2.纤维母细胞增生,有时小血管也增生。慢性炎症出现这些变化的机制尚在研究中,但有些介质可能起一定作用。体外实验证明,血小板衍生的生长因子(PDGF)和纤维粘连蛋白(FN)分解产物对纤维母细胞有趋化作用。此外,血小板衍生的生长因子、巨噬细胞和淋巴细胞的衍生因子都能在体外刺激纤维母细胞增生并产生大量胶原。因此,慢性炎症反应常伴有明显的瘢痕形成,造成肠道等的狭窄和浆膜面粘连。巨噬细胞衍生的可溶性因子可在活体内刺激血管新生。
3.局部组织的某些特殊成分如炎症灶的被覆上皮、腺上皮及其他实质细胞也可发生明显增生。
二、慢性肉芽肿性炎症
以在炎症局部形成主要由巨噬细胞增生构成的境界清楚的结节状病灶为特征的慢性炎症,称为慢性肉芽肿性炎症(chronic granulomatous inflammation)。结节较小,直径一般为0.5~2mm。这是一种特殊类型的慢性炎症。不同的病因可以引起形态不同的肉芽肿,因此病理学可根据典型的肉芽肿形态特点作出病因诊断,如见到结核性肉芽肿(结核结节)的形态结构就能诊断结核病。
1.慢性肉芽肿性炎症的常见病因
(1)某些细菌感染,其经典的例子为结核病、麻风和梅素。
(2)真菌和寄生虫感染,如组织胞浆菌病和血吸虫病。
(3)异物,如手术缝线、石棉和滑石粉等。
(4)原因不明,如肉样瘤病(sarcoidosis)。
2.形成肉芽肿的条件
(1)病原体(如结核杆菌)或异物(矿物油)不能被消化,刺激长期存在,造成慢性炎症。
(2)实验证明,刺激物所引起的细胞介导免疫反应在诱发慢性肉芽肿炎症中具有重要作用。体外实验证实,T细胞产物—淋巴因子可增强单核细胞向多核巨细胞转化。
3.肉芽肿的组成以结核结节为例,从结节中心向外,肉芽肿的成分依次为:
(1)干酪样坏死:典型结核结节的中央为干酪样坏死,内含坏死的组织细胞和白细胞,还有结核杆菌。结核结节中心的坏死可能是细胞介导免疫反应的结果。
(2)类上皮细胞(epithelioid cell):干酪样坏死灶周围可见大量胞体较大、境界不清的细胞。这些细胞的胞核呈圆形或卵圆形,染色质少,甚至可呈空泡状,核内可有1~2个核仁,胞浆丰富,染成浅红色。根据其形态与上皮细胞相似,故称类上皮细胞。
现在认为,在受一些不能被消化的细菌或其他抗原物质的长期刺激下,巨噬细胞就可转化为类上皮细胞,并发生剧烈的变化:①核内常染色质增多,说明DNA激活;②核仁增大靠近核膜,说明rRNA合成加强;③胞浆内富于内质网、核蛋白体及高尔基器和各种空泡,说明细胞器和酶的合成增强、分泌活跃;④线粒体、滑面内质网和溶酶体成分明显增多,因而胞浆成分扩展,细胞呈类似上皮细胞样紧密排列;⑤细胞膜Fc受体和C3b受体大大减少,吞噬功能随之明显降低。看来,类上皮细胞具有向细胞外分泌的功能,而不是吞噬作用。
经转化而形成的类上皮细胞的功能还不明确。但是他们可能通过分泌一些化学物质而杀伤细胞周围的细菌;同时在宿主健康组织与细菌之间组成一条隔离带。巨噬细胞在吞噬异物的过程中不会转化为类上皮细胞,若进入炎症灶的巨噬细胞过多,则有可能转化为类上皮细胞。
(3)多核巨细胞(multinucleated giant cell);在类上皮细胞之间散在多核巨细胞,结核结节之多核巨细胞又称为Langhans巨细胞。这种巨细胞体积很大,直径达40~50μm。胞核形态与类上皮细胞相似,数目可达几十个,甚至百余个,排列在细胞周边部呈马蹄形或环形,胞浆丰富。Langhans巨细胞系由类上皮细胞融合而成;类上皮细胞首先伸出胞浆突起,然后胞体相互靠近,最后经胞浆突起的融合使类上皮细胞融合在一起形成多核巨细胞。但融合的机制尚有待阑明。
多核巨细胞还常见于不易被消化的较大的异物(如手术缝线、石棉纤维等)和代谢产物(如痛风的尿酸盐结晶)周围。许多巨噬细胞围绕在刺激物周围并互相融合,形成由多数巨噬细胞质膜包围刺激物的巨大吞噬体,胞核散在分布于巨细胞胞浆中(图5-14)。这种多核巨细胞称为异物巨细胞(foreign body giant cell),多见于异物刺激引起的慢性肉芽肿性炎症。
多核巨细胞移动的速度十分缓慢,约每分钟0.04~1μm,而且随细胞核数目增多,移动速度更慢。巨噬细胞融合成多核巨细胞后,其胞膜的Fc和C3b受体数目也大大减少。因此,多核巨细胞的功能与类上皮细胞相似,可能分泌一些杀伤生物性病原体的物质并参与组成隔离宿主组织与细菌的屏障结构。
类上皮细胞和多核巨细胞均来源于巨噬细胞。若慢性肉芽肿性炎症的发生与细胞介导免疫有关,则巨噬细胞在局部集中还与激活的T细胞分泌的淋巴因子—巨噬细胞趋化因子(MCF)及巨噬细胞游走抑制因子(MIF)的作用有关。而且这些巨噬细胞还受巨噬细胞激活因子(MAF)的作用,提高其吞噬活性。
(4)淋巴细胞:在类上皮细胞周围可见大量淋巴细胞浸润。
(5)纤维母细胞: 结核结节周边有纤维母细胞及胶原纤维分布。
慢性炎症除一般性和特殊性表现形式外,有时还可形成炎性息肉和炎性假瘤。炎性息肉(inflammatory polyp)是在致炎因子的长期刺激下,局部粘膜上皮和腺体及肉芽组织增生而形成的突出于粘膜表面的肉芽肿块,常见于鼻粘膜和宫颈。炎性假瘤(inflammatory pseudotumor)是由组织的炎性增生形成的一个境界清楚的肿瘤样团块,常发生于眼眶和肺。
图5-14 异物巨细胞形成模式图
A.几个单核细胞(1)包围较大的异物(2)
B.单核细胞围绕异物形成多核巨细胞(1),异物位于吞噬体样袋(2)中
C.酸性粘多糖(1)被分泌进入吞噬体样袋
D.多数电子致密物质(1)沉着于多糖基质以加强对异物的包围
第六章 遗传与疾病
50年代以前,病理学所涉及的与遗传有密切关系的疾病,主要是对部分先天畸形进行了形态描述。有关遗传性疾病和先天畸形的现代丰富知识是近40年来积累的。随着染色体显示技术的进步,染色体的主要化学物质核糖核酸分子结构的揭晓以及关于越来越多的基因定位和功能的阑明,使遗传和疾病的研究进入了现代医学生物新领域,对于遗传性疾病和先天畸形的本质和病因的了解达到了新的水平。原则上,在遗传和疾病的关系上,人类所有疾病都具有遗传影响和背景,但只有在大约12%的疾病中,遗传因素起主要作用。
细胞遗传物质的受损,主要表现为基因突变(gene mutation)和染色体畸变(chromosomal aberration)。基因突变在分子上的表现,包括基因组核苷酸顺序的增加、减少或替换。因为一个基因的长度平均仅几个纳米(nm),所以在单一基因突变时,染色体核型往往无异常可见。但采用现代分子杂交技术结合染色体分带技术,有时可测出特定染色体特定部位上的核苷酸异常。染色体畸变均可用标准核型分析或分带技术、高分辨技术识别出来。由人体生殖细胞或受精卵的遗传物质发生改变而引起的疾病,可从亲代传至后代,称为遗传性疾病,这类疾病通常可分为三大类,即染色体病(chromosomal disease)、单基因遗传病(monogenetic or single -gene disease)和多基因遗传病(polygenetic or multigene disorder),近年又有线粒体遗传病(mitochondrial disease)。出生时即存在的人体形态或结构上的异常,称为先天畸形,属出生缺陷。这类疾病是一组来源不同的疾病,其中相当一部分与单个基因或染色体异常有关;有时是由已知环境因素(如致畸原)作用于胚胎发育过程中,造成体细胞损伤所致,后者不遗传。根据不同国家的大量统计表明,在世界范围,新生儿中至少2%有明显的先天异常,大约有1%的单基因病和0.5%的染色体病。临床工作中大约3%~4%有出生缺陷的病人可找到染色体核型异常,通过家系调查、生化检查等,有15%~20%的出生缺陷病人可判定属于其他各类遗传病。染色体病虽仅占新生儿出生缺陷的很小部分,但早期妊娠自发流产胎儿,经细胞遗传学分析检查,却有近2/3发现染色体异常,说明染色体畸变常是致死性的,仅少数出生并存活。同时也须指出,有染色体核型异常的人,可不出现能识别的表型异常。根据McKusik提供的资料表明,单基因遗传性状的发现,由1971年的1876种增至1994年的6678种。在这段期间国际上以平均每年发现100多种新单基因遗传病的速度在进展,也反映出遗传病带给医学、社会和家庭问题的严重性。在人类的基因组内约有3.3×109 碱基对,如果按每千碱基长度为一个基因,可能有2×106个基因,估计其中50000~100000为有活性的基因,因此目前所发现的单基因遗传病,仅占结构基因的百分之几。随着方法学的进步,还将发现和认识更多的遗传性疾病。
在肿瘤细胞,特别是在恶性肿瘤细胞,大多数可观察到基因或染色体异常。通常认为肿瘤是体细胞突变的结果。由于体细胞突变不影响生殖细胞,故细胞突变的遗传特征不会传给个体的下一代。但突变的细胞会形成一团基因型与体内其他细胞不同的细胞群。至于肿瘤疾患和宿主的遗传因素的关系,不少学者认为大多数肿瘤应属于多基因遗传病范畴。Knudson和Strong根据肿瘤的遗传流行病学特点,提出关于肿瘤发生的两次突变假说(two mutation hypothesis),即肿瘤必须经过二次以或二次以上的细胞突变才能形成。具有癌易感性素质的患者,第一次突变可能发生或存在于生殖细胞,而第二次突变则发生于体细胞。必须有第二次突变才能使敏感了的细胞发生转化。关于肿瘤患者的遗传因素影响在肿瘤章中途述。
第一节 遗传病
遣传性疾病通常具有垂直传递的特征,可由亲代传至后代。有时突变发生在配子形成(gametogenesis)时期,因此父母双方都没有这类缺陷,这类患者成为起始性突变者,可能成为后代子孙患病的祖先。有些基因突变或染色体畸变是致死性的或明显降低生殖力,以致这种缺陷不能传递。多基因型传递的遗传疾病,可能经长期一再地与正常个体婚配,而冲淡突变基因甚至消失。遗传性疾病常具有先天性、终生性和家族性的特点。但先天性疾病并不都是遗传病,在胎儿发育过程中,由于环境因素或母体条件的影响,可出现非遗传性先天性疾病。遗传性疾病也常表现为晚发,一些致病基因的作用仅在个体发育达到一定年龄后才表现出来。尤其是那些由遗传基因和环境条件两种因素支配着表现型的遗传性疾病。因此,晚发的疾病仍可以是遗传性疾病。遗传性疾病常可表现为家族性,这是因为同一家系中的成员可共同具有某一致病基因,但同一家系的成员也处于相似的生活条件和环境中,由相似环境条件所引起的非遗传性疾病,有时也具有家族性。一些遗传性疾病(隐性遗传病)仅在基因型为纯合子状态下才表现出来,因而发病的概率小,相似于散发性疾病。
作为遗传病的发病基础,近年来的认识越加深入。已往知之较多的是染色体畸变,畸变处出现染色体遗传物质的得失、断裂或位置改变而使机体出现病状。另一方面是基因突变,是编码基因的碱基序列发生变化,随之表达和表达产物异常。这种基因突变引起的疾病,随该基因所在染色体连锁、交换,按孟德尔的自由组合及分离律传递给下一代,称为孟德尔式遗传病。人类线粒体DNA是细胞核外DNA,它的DNA突变也可致线粒体功能异常出现疾病,总称线粒体遗传病。线粒体遗传病是由卵细胞胞浆内突变的线粒体DNA传递给下一代的。因此表现为母系遗传。家系中可见男女均可患病,但男患者后代再无患者。近年来人类基因研究发现,父源和母源染色体上的某些基因不能互相替代。基因由父方传给子女和由母方传递给子女时,常有不同的表现。这是由于基因在生殖细胞(精子和卵子)分化过程中受到不同的修饰。这种现象称为遗传印记(genetic mprinting)。目前认为遗传印记的分子机制主要是DNA的甲基化。已知甲基化可以抑制基因的表达。一些基因在精子和卵子的甲基化状态不同,因此使该基因的表达况有所不同。高度甲基化的被印记的基因可以不表达或表达程度很低,因而可影响到疾病的发生、外显率、表现度,甚至遗传方式。存在这种基因外的修饰现象,也可以解释何以来自父方和母方的某些基因不能互相替代,也可以说明一些孟德尔单基因遗传病却表现出非孟德尔遗传方式。这是又被遗传印记修饰、改变的结果。在遗传病基因研究上,还发现了一类疾病存在有三联序列的高度重复现象。这类疾病有脊髓延髓肌萎缩(Kennedy病,CAG三联体重复)、脆性X综合征(CGG三联体重复)、强直性肌萎缩(GCT三联体重复)。在脆性X综合征的CGG重复序列研究中,发现正常人CGG序列重复6~54次,峰值为29,且各种族间无显著差异。正常男性传递者和女性携带者此序列的重复次数可分别增加到82次及83~90次,这种状态可认为属前突变。女性携带者在传递给下代男性患者而发病时,男性患者的CGG重复次数可高达250~4000次。这种三联体重复序列逐渐增加的突变形式,称为不稳定动态突变。由于这种不稳定的动态突变致使一些疾病发生。除上述所举三种疾病外,1993年3月又报道了Huntington舞蹈病,也是CAG三联体重复,但部位在基因前半部与延髓脊髓肌萎缩在后半部不同。遗传病的研究是当代学术进展最快的领域之一,对其发病基础的知识日益丰富。
一、染色体病
染色体病是由于染色体数目或结构异常而发生的疾病。染色体数目异常比结构异常更为常见。
1.数目性染色体畸变染色体的数目异常可表现为非整倍体(aneuploid),即其数目并非单倍体(haploid)的整倍数,在数目上出现多或少于整倍体,如45或47条染色体。还可表现为多倍体(polyploid),即染色体数目整倍多于单倍体,如三倍体(triploid)69条,四倍体(tetraploid)92条。常见的非整倍体患者是某对染色体不是2条而出现3条,称为三体综合征(trisomic syndrome)。如果某对染色体缺少1条,则称为单体综合征(monosomic syndrome)。多倍体患者很少见,可见于肿瘤细胞和流产胎儿。
染色体数目异常几乎全是减数分裂不分离(non disjunction)或分裂后期迟延(anaphase lag)的结果。在第一或第二次减数分裂时期,由于两条同源染色体未能分开,而造成子代细胞染色体数目或多或少。
2.结构性染色体畸变这种畸变是在细胞分裂过程中曾有染色体断裂所致。常见的结构异常有缺失、环状染色体、易位、重复、倒位和等臂染色体(图6-1)。
(1)缺失:指染色体丢失一段。即染色体一处断裂,其无着丝粒的一端常丢失,成为末端缺失;染色体两处断裂,可造成中间段的丢失,为中间缺失。由于遗传基因随染色体断片而丢失,可造成该个体患严重的多发缺陷。
(2)环状染色体:一条染色体的两端发生断裂,两侧末端片段丢失,断端相互连接形成环状染色体。
(3)易位:当两条非同源染色体同时发生断裂时,断落片段由一条染色体移至另一条染色体的断端上,形成易位染色体。易位可以是平衡性的,也可以是不平衡性的。在易位发生过程中,可造成染色体的点缺失,基因断裂损伤或位置效应,由此产生表型异常,此种称为不平衡易位(unbalanced translocation)。没有遗传物质的得失者,表型正常,称为平衡性易位(balanced translocation)。这两种易位很难从常规染色体检查中区分。
(4)重复:是指一条染色体的断片移至同源染色体的相应部位上,造成该同源染色体此段重复。
(5)倒位;是指一条染色体两处断裂,中间的片段倒转180°后,再与两断端连接起来,形成倒位,这种倒位可以发生在两臂间,也可发生在臂内。
(6)等臂染色体:是着丝粒分裂异常所致。正常应是纵裂分开,如果横裂分开,短臂与短臂,或长臂与长臂相接,各形成等臂染色体。
图6-1 人类染色体几种结构畸变的模式图
由上可见,染色体断裂是结构重排、结构异常的重要前提。没有先发生断裂,就没有结构的异常。断裂本是常见的生理现象,减数分裂或有丝分裂中均可发生。用姊妹染色单体交换检查方法可查出。尽管断裂经常发生,但显然大部分断裂可“自愈”。而不引起可查觉的染色体结构变化。因此,断裂后所致染色体的结构畸变的具体原因和机制尚不很清楚。但已了解到有一些因素可引起断裂增加,如射线照射、病毒感染、药物和环境因素等。几种称为染色体断裂综合征(chromosome breakage syndrome)的疾病,如毛细血管扩张性共济失调症(ataxia teleangiectatica)、Bloom综合征、着色性干皮症(xeroderma pigmentosa)和Fanconi贫血等,病人的染色体裂隙和断裂发生率增高。
3.几种染色体病举例
(1)Down综合征:是最常见的常染色体病,其染色体异常在21三体,主要核型为三体型,少数为易位型或嵌合型。表型特征有智力低下、伸舌、鼻梁低平、眼裂上斜、小耳、小颌、枕平、内眦敖皮、颈短及肌张力减低等,常伴有先天性心脏发育缺陷。患者急性淋巴性和粒细胞性白血病的发生率增高。发病率随母龄增长而增高(图6-2)。
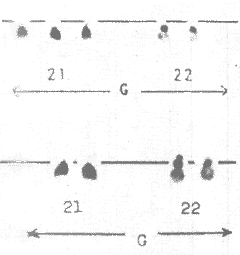
图6-2 21三体核型与正常核型比较
上图 21三体(Down)综合征下图 正常核型
(2)Edwards综合征:染色体异常为18三体。表型特征有智力低下、小头、前额窄、枕部突、小颌且张口范围小,腭弓高窄、低位耳、肾畸形(图6-3)、肌张力增高及手紧握等(图6-4)。
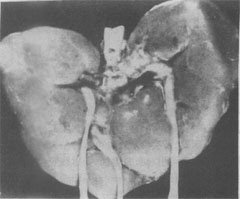
图6-3 18三体(Edwards)综合征的畸形肾
图示马蹄状融合肾及额外输尿管
图6-4 (Edwards)综合征的肌张力增高
图示手紧握及特殊拳方式
(3)Patau综合征:染色体异常为13三体。表型特征中有中枢神经系统发育缺陷,呈前脑无裂畸形(holoprosencephaly)(图6-5)。前额小呈斜坡样,头皮后顶部常有缺损。视网膜发育不良,切片镜下观察可见菊形团形成。严重智力低下,唇裂及腭裂、多指等。中性粒细胞核上有过多的突起。
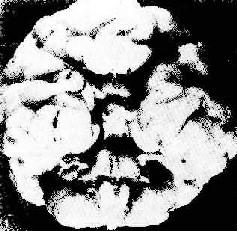
图6-5 13三体(Patau)综合征脑畸形
图示前脑无裂
(4)猫叫综合征(cri du chat syndrome):染色体异常为5号染色体短臂部分或全部缺失。表型特征有:婴儿时期哭声似猫叫,因而得名,可有喉器小和会厌小等喉部发育异常。严重智力低下,小头、圆形脸、眼距宽、眼裂下斜、耳位低、内眦赘皮等。
(5)Turner综合征:这是唯一已知的性染色体单体病(sex chromosome monosomy)。也称先天性卵巢发育不全症。典型核型是45,X。表型特征有:身材矮小,颈蹼及指、趾背部浮肿,为胎儿期淋巴水肿的残迹。后发际低、盾状胸、肘外翻,卵巢发育不全、原发闭经,性器官幼稚型(图6-6)。

图6-6 Turner综合征的一种核型 46,X,r(X)
由于一条X染色体呈环形,末端片段有丢失,影响卵巢发育
(6)Klinefelter综合征:又称先天性睾丸发育不全症。典型的核型是47,XXY。表型特征有睾丸发育不全。身材修长,乃足跟至趾骨间的距离增长所致。男子乳腺发育、阴毛呈女性型分布,阴茎及睾丸均小。严重者伴有智力迟钝、隐睾及尿道下裂等(图6-7)。

图6-7 Klinefelter综合征的核型47,XXY
由于增加一条X染色体,影响睾丸发育
染色体核型分析时,有时可见到一个患者有两种不同的核型。如果这两种不同核型的细胞起源于单一合子(zygote),则这种由两种或两种以上细胞组成的个体称为嵌合体(mosaicism)。起源于一个以上的合子时,称为异源嵌合体(chimera)。如Klinefelter综合征常可见46,XX/47,XXX/48,XXXY等嵌合体。Dwn综合征也可见46,XY(XX)/47,XY(XX),+21的嵌合体。异常核型细胞所占比例少者症状较轻。
二、单基因遗传病
起源于单一(对)基因的突变。单基因遗传病的传递方式是按孟德尔法则(Mendelian law)传至后代的。新突变所致的患者可无家族史。目前已知的单基因遗传病有3000余种。根据突变基因所在的位点和性状的不同,而区分为下列不同类型。
1.致病基因可以是生殖细胞发生突变而新产生,也可以是由双亲任何一方遗传而来的。此种患者的子女发病的概率相同,均为1/2。此种患者的异常性状表达程度可不尽相同。在某些情况下,显性基因性状表达极其轻微,甚至临床不能查出,这种情况称为失显(non penetrance)。由于外显不完全,在家系分析时可见到中间一代人未患病的隔代遗传系谱,这种现象又称不规则外显(irreqular dominance)。还有一些常染色体显性遗传病,在病情表现上可有明显的轻重差异,纯合子患者病情严重,杂合子患者病情轻,这种情况称不完全外显(incomplete dominance)。常染色体显性遗传病常见者有Marfan综合征、Ehlers-Danlos综合征、先天性软骨发育不全、多囊肾、结节性硬化、Huntington舞蹈病、家族性高胆固醇血症、神经纤维瘤病、肠息肉病以及视网膜母细胞瘤等。
2.常染色体隐性遗传病(autosomal recessive disorder)致病基因在常染色体上,基因性状是隐性的,即只有纯合子时才显示病状。此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。子代有1/4的概率患病,子女患病概率均等。许多遗传代谢异常的疾病,属常染色体隐性遗传病。按照“一个基因、一个酶”(one gene one enzyme)或“一个顺反子、一个多肽”(one cistron one polypeptide)的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症、脂质贮积症、粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆状核变性(Wilson病)及半乳糖血症等。
3.性连锁遗传病(sex-linked disorder)以隐性遗传病为多见。致病基因在X染色体上,性状是隐性的,女性大多只是携带者,这类女性携带者与正常男性婚配,子代中的男性有1/2是概率患病,女性不发病,但有1/2的概率是携带者。男性患者与正常女性婚配,子代中男性正常,女性都是携带者。因此X连锁隐性遗传在患病系中常表现为女性携带,男性患病。男性的致病基因只能随着X染色体传给女儿,不能传给儿子,称为交叉遗传。这类常见的疾病有血友病A、假性肥大性肌营养不良症(Duchenne肌营养不良),红绿色盲等。其中红绿色盲如女性携带者和男性患者婚配,子代中的男性有1/2的概率患病,而女性可有1/2的概率患病及1/2概率为携带者。
X连锁显性遗传病病种较少,有抗维生素D性佝偻病等。这类病女性发病率高,这是由于女性有两条X染色体,获得这一显性致病基因的概率高之故,但病情较男性轻。男性患者病情重,他的全部女儿都将患病。
Y连锁遗传病的特点是男性传递给儿子,女性不发病。因Y染色体上主要有男性决定因子方面的基因,其他基因很少,故Y连锁遗传病极少见。
三、多基因遗传病
人类许多生理特征如身高、体重、血压和肤色深浅,毛发疏密等,是受多项基因决定的。多基因遗传病是由两对以上微效基因共同作用造成的,无显性和隐性之分。每对基因作用较小,但有积累效应。在发病时还常受环境因素的影响,故也称多因子遗传(multifactorial inheritance)。这类疾病中遗传因素所起的作用程度不同,按其程度大小以百分率(%)来表示,称为遗传度。环境因素影响越大,遗传度越低。唇裂、腭裂、高血压、糖尿病、躁狂抑郁症、类风湿性关节炎及先天性心脏病等,均有多基因遗传基础,并各自有其遗传度。
要确定某一疾病为多基因遗传病,是比较困难的,首先要除外染色体病和单基因遗传病,还要进行较为周密的家系调查。
第二节 先天畸形
一、畸形学基本概念
器官或组织的体积、形态、部位或结构的异常或缺陷,称为畸形(malformation)。先天畸形是指出生时即存在的形态或结构上的异常。有单发畸形(如唇裂、多指等)和多发畸形之分。许多种多发畸形是在某一原因作用下特异地组合而发生的,成为畸形综合征,目前已经识别诊断的畸形综合征已达250余种。能引起先天畸形发生的作用物,有化学性、物理性或生物性等物,称为致畸因子或致畸原(teratogen)。目前已发现多种多样的致畸形因子,它们的有害影响能在动物实验中证实,仅少数可确证对人类有影响。因此,只能视之为对人类有很大的潜在危险,应予重视。
在致畸因子作用下,是否发生畸形,结果如何,还取决于下列一些因素:
1.孕妇对致畸因子的感受性,在个体之间存在着差异。
2.胎儿发育的不同阶段,对致畸因子的感受性不同,大多数致畸因子有其特定的作用阶段。
3.致畸因子的作用机制有所不同,例如有些致畸药物可抑制酶或受体的活性,有些是干扰分裂时期纺锤体的形成,还有的封闭能源并抑制能量的产生,进而抑制正常形态发生所需的代谢过程。许多药物和病毒对某种组织、器官有特别亲和性,故特别易侵及某种组织和器官,如所谓亲神经性或亲心脏性等,它们会损伤一些特定的器官,影响其发育。
4.致畸因子的损伤与剂量有关,通常是剂量越大,毒性越大。理论上讲,应该有安全剂量。但实际上,由于致畸过程具有多方面的决定因素,难以一概而论,故已经确定的致畸剂在妊娠期间应绝对避免,因为尤其是对个体来讲,难于判定绝对安全剂量。
5.致畸因子的作用后果,包括胎儿死亡、生长发育延迟、畸形或功能缺陷。究竟出现何种后果,则取决于致畸因子,母体及胎儿胎盘的相互作用如何。
二、胚胎的发育和畸形的形成
前已提及,胎儿在发育的不同阶段,对于致畸因子的感受性不同。畸胎的发生,特别要追溯到胚胎发育的早期阶段。
1.植入前期(preimplantation stage) 从受精到第6天。正常的受精卵(合子,zygote),头3天在输卵管中发育。受精后30小时才完成双细胞阶段,以后分裂速度增加,发育成桑椹球体。受精卵含有成人机体的所有基因成分,但它们绝大部分处于不激活状态,在受精卵进入发育的分裂阶段,激活的是那些与复制、生长、细胞间相互作用有关的基因。如果受精卵带有异常的结构基因或者不正常地激活了某些基因,将会导致早期胚胎细胞死亡。如果将发育成2~4个细胞的分裂球(blastomere)从细胞间加以分开,则可形成同卵孪生儿。由于分裂球的组成细胞均属潜力均等的全能细胞,因此理论上讲,如果丢失一个细胞可能并不带来严重后果。但事实上也正由于它们彼此一致,一个细胞带有致死性的基因,可能其他细胞也带有同样的致死性基因。激活这种基因必然导致死亡。同时也正是由于这些细胞的彼此一致,一旦暴露于不利的外源性影响下,有害因子对所有细胞会产生同样伤害,因而造成胚胎死亡。因此,通常认为有害环境因子对植入前期胚胎发挥着“全或无”的影响,也就是说或者胚胎死亡,或者存活,存活即意味着未受损伤,不会致畸。
2.植入期(implantation stage) 从第1周末至第2周末。胚泡到达宫腔内膜并开始植入。在8~10天阶段,如果受到损伤,有可能造成分裂中细胞的彼此不完全分离,因而形成联胎(double monsters)。联胎可以是对称性的,也可以是不对称性的,对称性联胎也可以说是分离不完全的孪生儿,它们在不同部位仍相连在一起,例如头部、胸部或臀部。不对称性联胎是指一个孪生儿发育良好,而另一个则发育不良或仅有残迹。发育不良者常表现异常,或在体外与发育良好者相连,或者存在于发育良好的体内,而成为“胎中胎”。一些先天性畸胎瘤,特别是骶尾部的,实际上是不对称性的联胎。
3.三胚层期至第2个月末在植入期,已开始有内、外胚层的区分。从第3周至第1个月末,两胚层之间又出现中胚层,各层并开始分化形成各原始的组织器官系统。细胞分化、迁移是这一阶段的特点。畸胎的发生多起始于胚胎这一发育阶段。第2个月各原始的器官系统逐步发育成形,各种形态发生上的异常以及广泛而严重的畸形,多在这阶段致成。因此胚胎的两周至12周,是致畸因子损伤而造成先天畸形的关键时期。
4.胚胎3个月至产前从胚胎第3个月,各系统在已建立的相互联系条件下,受神经系统的支配,开始了原始的胎儿活动,主要器官系统趋于完整。此时,相对而言,对于致畸因子的作用有了抵抗力。但器官系统仍在不断完善和功能适应。但已分化器官的功能成熟程度不同,因此也不能完全排除致畸因子的不良影响。从第4个月开始,胎儿进入迅速生长阶段,胎儿生长受阻而造成的变形,有可能在这一时期形成。子宫内的羊膜带(amniotic band),甚至子宫壁本身,都可能带给胎儿损伤过程。
三、先天畸形的成因
1.遗传因素形态发生是受遗传因素控制的。先天畸形的发生绝大多数与遗传有关,如染色体畸变、单基因遗传、多基因遗传等。
2.环境因素畸胎学的研究不断发现环境中的一些物质可以造成胎儿畸形。在分析某个致畸因子的致畸风险时,应该考虑该致畸因子的作用强弱、剂量大小、持续时间、孕母的遗传背景以及当时的孕龄等。常见的致畸因子有:
(1)辐射:其中离子电磁辐射有较强的致畸作用,包括α、β、γ和X射线,其致畸作用与各射线的穿透力有关。日常,人们都或多或少地接触射线,有的人还因居住环境或职业关系可能接触更多的射线,但对其致畸作用要具体分析。非电离性辐射,包括短波、微波及紫外线等,其致畸作用较弱。其中紫外线对DNA修复机制有缺陷的患者是一种致突变因子。
(2)X线:一般说,诊断性X线检查,对胎儿的危害不大,但也与照射部位有关,例如胆囊造影只使胎儿接受0.000006Sv(注:Sv是剂量当量单位,1Sv=100rem);而钠灌肠造影检查,由于直接照射骨盆,则可使胎儿接受0.0035Sv。另外,治疗剂量要大得多。治疗脑肿瘤、乳腺癌(单侧)均可使胎儿接受0.09Sv,治疗肺癌可接受0.25Sv。所以,用于治疗的X线有致畸的危险。
(3)同位素:各种组织对不同的同位素吸收量不同,例如口服5mci的131I,甲状腺可接受100Sv,而性腺只有0.12Sv;口服4mci 32P,有1.0Sv进入骨髓、肝、脾,而其他部位则不过0.1Sv。另外,胎儿对同位素的吸收程度还与胎龄有关,例如胎儿在第10周,从循环中结合的碘比母亲甲状腺结合的还要多。因此孕妇必须用放射性碘进行诊断时,应在胎龄第5~6周之前进行,即在胎儿甲状腺分化之前完成。
X线、同位素、还有其他外源性离子辐射对分裂细胞的影响,包括杀伤细胞、抑制有丝分裂、改变细胞的正常迁移和彼此联系,以及造成染色体畸变和基因突变等。植入前期,大剂量照射可导致胚胎死亡,这是由于致死性染色体畸变或细胞分化受损所致。胚胎两周后的任何时期接受超过1.0Sv的射线,均可造成器官畸形或生长受阻,其中以中枢神经系统最为敏感,0.25Sv时就可发生小头畸形,智力低下,有实验证明,不伴有神经系统导常的其他畸形,则可谓并非由辐射引起,此剂量目前为大多数所接受的致畸剂量标准,胎儿接受0.25Sv以上的照射,有致畸危险。美国学者大多数以0.1Sv作为阈值来推断致畸危险。有时尽管不发生畸形,但轻微的损伤可引起智商降低,将来癌变也很难排除与辐射的关系。
3.病毒和感染可致畸的病毒感染有风疹、巨细胞病毒感染、水痘及单纯疱疹B。其他病毒感染的致畸问题,目前尚无足够的证据。畸形儿是否与感染有关,可通过检查IgM来判定,受感染的新生儿IgM上升。也可进行病原基因检测。
(1)风疹(rubella):前四周受感染,致畸危险为61%,5~8周时为26%,9~10周时为6%。风疹综合征的畸形包括眼、耳、心、造血系统、中枢神经系统、肺、肝和骨。神经系统损伤如小头、智力低下,眼的缺陷方面有视网膜病变、白内障、青光眼、小眼球等。
(2)水痘(varicella):致畸危害只是在胚胎前16周或产前4天之内发生。前16周感染,可致畸,包括眼的缺陷如白内障、小眼球、视神经萎缩以及脑损伤和肢体发育不全等。分娩前4天孕妇感染水痘,有20%的新生儿死亡。
(3)巨细胞病毒感染(cytomegalovirus infection):大约有10%的感染胎儿出现神经系统损伤。可有智力减低、耳聋。还可有低出生体重、小头、肝脾肿大及血小板减少性紫癜、视神经萎缩等。进行病毒分离和尿中脱落细胞查找病毒包涵体,均可证明感染的存在。
(4)弓形虫感染(toxoplasma infection):主要表现为眼的疾患,90%有脉络膜炎,50%~60%有癫痫、小头和脑积水。即使感染得到控制,也常遗留眼或脑的损害。
4.化学药物苯妥英钠可致成轻、中度生长发育及智力障碍。妊娠期服用此药,胎儿致畸危险为1/10~1/3。三甲双酮(抗癫痫药)会造成胎儿智力低下、发育缓慢、面部发育不良、唇腭裂、房间隔缺损及两性畸形。孕妇服用丙酮苄羟香豆素(抗凝剂),可引起胎儿软骨发育不良,多表现为低出生体重及智力低下,中枢神经系统有异常。早孕妇女服用此药,胎儿约1/3发生畸形。氨基蝶呤(抗叶酸剂),曾被用于进行人工流产,未流产者可造成胎儿多发畸形,其衍生物氨甲蝶呤有同样致畸作用。在抗生素药物方面,四环素可与骨盐形成复合物,使牙变色;链霉素可致胎儿耳聋。激素方面,雄激素去甲睾丸酮衍生物用于避孕,可使女胎男性化,对男胎无不利影响;雌激素复合物氯菧酚胺可致畸,使非整倍体增加,可出现椎骨、心脏、肢体的畸形;皮质激素有诱发缺肢、先天性心脏病的报道;胰岛素可使神经管缺陷增多,还可造成先天性心脏病和肢体缺陷。
四、先天畸形的形成方式和类型
1.先天畸形的形成方式
(1)胚胎组织形成不良:在遗传和环境致畸因素的影响下,使胚胎本身有着内的缺陷,因而造成组织器官形成不良,产生畸形。可单发或多发。
(2)变形(deformation):胚胎本身原无缺陷,各组织、器官早期发育原本正常,只是由于受到外来机械力作用,使原来正常发育的组织、器官受压变形,出现畸形。来自母亲的机械压力有:双角子宫、子宫肌瘤、骨盆狭小等;属于胎儿方面的有:过早入盆、胎位不正和羊水过少、多胎、胎儿过大等。
(3)胚胎组织或胎儿的发育过程受到外来作用的阻断,造成畸形。最明显的例子是近年发现的羊膜带形成及其影响作用。羊膜带可因羊膜在妊娠早期破裂而形成,由于羊膜带与胎儿体表的粘连或缠绕,使早期发育着的胚胎或胎儿的组织、器官发育阻滞和破坏,产生畸形,形成羊膜带阻断症。
2.先天畸形类型先天畸形的种类很多,人类可识别的多发畸形已有300多种,这些不同表现的畸形,可按其形成方式归纳为五种主要类型来识别。
(1)综合征(syndrome):所谓综合征是指一群或几种畸形,经常共同出现在一个个体中。综合征常起因于同一病因,如21三体可形成Down综合征,风疹感染可引起风疹综合征。
(2)联合征(association):指一群或几种畸形常伴同在一起,出现在同一个体中。但不如综合征那样恒定,也不是偶然的巧合,这样一组畸形称为联合征。联合征可能系不同病因所致。如VATER联合征,由脊柱(V)、肛门(A)、气管(T)、食管(E)、肾(R)等畸形联合而成。
(3)变形症(deformation):指外来机械力作用引起的变形。常见的有踝内收、胫骨扭曲等。
(4)阻断症(disruption):指胚胎或胎儿本身没有内在缺陷,在发育中胎儿体外的某些因素如羊膜带或体内血栓形成等,使组织、器官的发育受阻或破坏,造成畸形。羊膜带常因妊娠前8周时羊水早破而形成,呈片状或带状。羊膜带产生的时间越早,危害越严重。大部分畸形出现在体表,即上述羊膜带阻断症。
(5)序列征(sequence):在胚胎发育中,在某种因素影响下,先产生一种畸形,由此畸形进一步导致相关组织、器官的一系列畸形。这一连串发生的畸形称为序列征。例如Robin-Pierre序列征,起始畸形为小下颌,因而舌被迫向后向上移位,致使腭板不能正常闭合,并使呼吸道受阻,出现一连串畸形。由单一组织发育不良形成的序列畸形,称为畸形序列征;由变形引起的序列畸形,称为变形序列征;由阻断引起的序列畸形,称为阻断序列征。
医学遗传学和畸胎学的研究,力图阐明畸形发生的原因和机制,并为预防畸形寻找途径。目前行之有效的方法有:卫生宣教,家系调查,遗传咨询,畸形及风险预测,生物化学检验,羊水和绒毛检查,基因检测和细胞遗传学分析以及临床B型超声波及胎儿镜的应用等,已取得一定效果,尚有待进一步发展和完善。
第七章 肿瘤
肿瘤(tumor,neoplasm)是一种常见病、多发病,其中恶性肿瘤是目前危害人类健康最严重的一类疾病。在欧美一些国家,恶性肿瘤的死亡率仅次于心血管系统疾病而居第二位。根据我国1992年卫生事业发展情况统计公报,城市地区居民恶性肿瘤死亡率居死因第一位,为123.92/10万,基中男性为151.08/10万,女性为95.12/10万。农村地区居民恶性肿瘤死亡率居第死因第二位,为101.39/10万。其中男性为125.79/10万,女性为78.85/10万。随着我国人口老龄化的进程,加之城市人口比例逐年增高,城镇工业生产迅速发展,环境污染日益严重,吸烟等不良生活习惯相当普遍,如果不采取积极的宣传教育措施,恶性肿瘤的危害性还将日益增加。我国最为常见和危害性严重的肿瘤为肺癌、鼻咽癌、食管癌、胃癌、大肠癌、肝癌、乳腺癌、宫颈癌、白血病及淋巴瘤等。特别是肺癌发生率近年来有明显的增加,值得重视。这些肿瘤的病因学、发病学及其防治,均应为我国肿瘤研究的重点。
第一节 概述
一、肿瘤的概念
肿瘤是机体在各种致瘤因素作用下,局部组织的细胞在基因水平上失掉了对其生长的正常调控,导致异常增生而形成的新生物。这种新生物常形成局部肿块,因而得名。
正常细胞转变为肿瘤细胞后就具有异常的形态、代谢和功能,并在不同程度上失去了分化成熟的能力。它生长旺盛,并具有相对的自主性,即使后来致瘤因素已不存在时,仍能持续性生长,不仅与机体不协调,而且有害无益。
机体在生理状态下以及在炎症、损伤修复时的病理状态下也常有组织、细胞的增生。但一般来说,这类增生有的属于正常新陈代谢所需的细胞更新;有的是针对一定刺激或损伤的适应性反应,皆为机体生存所需。再者,所增生的组织能分化成熟,并能恢复原来正常组织的结构和功能。而且这类增生是有一定限度的,一旦增生的原因消除后就不再继续增生。但肿瘤性增生却与此不同,二者有着本质上的区别。
根据肿瘤生物学特性及其对机体危害性的不同,一般分为良性和恶性肿瘤两大类。这种分类在肿瘤的诊断、治疗和判断预后上均有十分重要的意义。
二、肿瘤的一般形态和结构
1.肿瘤的肉眼观形态 在肉眼观上,肿瘤的形态多种多样,并可在一程度上反映肿瘤的良恶性。
肿瘤的数目和大小肿瘤的大小不一,通常一个,有时可为多个。小者极小甚至在显微镜下才能发现,如原位癌(carcinoma in situ);大者很大,可重达数公斤乃至数十公斤。一般说,肿瘤的大小与肿瘤的性质(良、恶性)、生长时间和发生部位有一定的关系。生长于体表或大的体腔(如腹腔)内的肿瘤有时可长得很大;生长于狭小腔道(如颅腔,椎管)内的肿瘤则一般较小。大的肿瘤通常生长缓慢,生长时间较长,且多为良性。恶性肿瘤生长迅速,短期内即可带来不良后果,故一般不致长得很大。
肿瘤的形状 肿瘤的形状多种多样,有乳头状、菜花状、绒毛状、蕈状、息肉状、结节状、分叶状、浸润性包块状、弥漫性肥厚状、溃疡状和囊状等(图7-1)。肿瘤形状上的差异一般与其发生部位、组织来源、生长方式和肿瘤的良恶性质密切相关。
肿瘤的颜色 一般肿瘤的切面多呈灰白或灰红色,但可因其含血量的多寡、有无变性、坏死、出血,以及是否含有色素等而呈现各种不同的色调。有时可从肿瘤的色泽大致推测其为何种肿瘤,如血管瘤多呈红色或暗红色,脂肪瘤呈黄色,黑素瘤多呈黑色,绿色瘤呈绿色等。
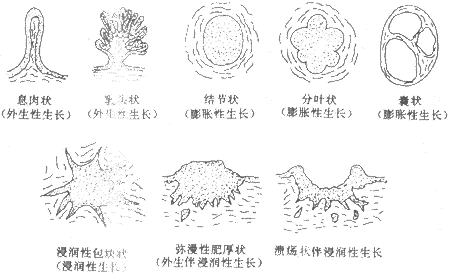
图7-1 肿瘤的外形和生长方式模式图
肿瘤的硬度肿瘤一般较其来源组织的硬度增大,其硬度与肿瘤的种类、瘤实质与间质的比例以及有无变性坏死等有关。如骨瘤很硬,脂肪瘤质软;实质多于间质的肿瘤一般较软,反之则较硬;瘤组织发生坏死时变软,有钙质沉着(钙化)或骨质形成(骨化)时则变硬。
2.肿瘤的组织结构 肿瘤的组织多种多样,但概而言之,任何一个肿瘤的组织成分都可概括为实质和间质两部分。
肿瘤的实质 肿瘤实质是肿瘤细胞的总称,是肿瘤的主要成分。肿瘤的生物学特点以及每种肿瘤的特殊性都是由肿瘤的实质决定的。机体内几乎任何组织都可发生肿瘤,因此肿瘤实质的形态也多种多样,通常根据肿瘤的实质形态来识别各种肿瘤的组织来源(histogenesis),进行肿瘤的分类、命名和组织学诊断;并根据其分化成熟程度和异型大小来确定肿瘤的良恶性。
肿瘤的间质 肿瘤的间质成分不具特异性,起着支持和营养肿瘤实质的作用,一般系由结缔组织和血管组成,有时还可有淋巴管。通常生长快的肿瘤,其间质血管多较丰富而结缔组织较少;生长缓慢的肿瘤,其间质血管则较少。此外,肿瘤间质内往往有或多或少的淋巴细胞等单个核细胞浸润,这是机体对肿瘤组织的免疫反应。近年来在肿瘤结缔组织间质中除见纤维母细胞外,尚出现肌纤维母细胞(myofibroblast)。此种细胞兼具纤维母细胞和平滑肌细胞的一些形态和功能特点,细胞呈梭形。电镜下,胞浆内除见丰富的粗面内质网外,在细胞膜下可见平滑肌微丝。此种细胞既能产生胶原纤维,又有收缩功能。由于此种细胞增生、收缩和形成胶原纤维包绕肿瘤细胞,可能对肿瘤细胞的浸润过程有所阻抑,并限制瘤细胞的活动和遏止瘤细胞侵入血管内或淋巴管内,从而减少播散机会。此外,此种细胞的增生还可解释临床上所见乳腺癌时的乳头回缩,食管癌及肠癌所致的管壁僵硬和狭窄。
第二节 肿瘤的异型性
肿瘤组织无论在细胞形态和组织结构上,都与其发源的正常组织有不同程度的差异,这种差异称为异型性(atypia)。肿瘤组织异型性的大小反映了肿瘤组织的成熟程度(即分化程度,在此指肿瘤的实质细胞与其来源的正常细胞和组织在形态和功能上的相似程度)。异型性小者,说明它和正常组织相似,肿瘤组织成熟程度高(分化程度高);异型性大者,表示瘤组织成熟程度低(分化程度低)。区别这种异型性的大小是诊断肿瘤,确定其良、恶性的主要组织学依据。恶性肿瘤常具有明显的异型性。
由未分化细胞构成的恶性肿瘤也称为间变性肿瘤。间变(anaplasia)一词的原意是指“退性发育”,即去分化,指已分化成熟的细胞和组织倒退分化,返回原始幼稚状态。现已知,绝大部分的未分化的恶性肿瘤起源于组织中的干细胞丧失了分化能力,而并非是已经分化的特异细胞去分化所致。在现代病理学中,间变指的是恶性肿瘤细胞的缺乏分化。间变性的肿瘤细胞具有明显的多形性(pleomorphism,即瘤细胞彼此在大小和形状上的变异)。因此,往往不能确定其组织来源。间变性肿瘤几乎都是高度恶性的肿瘤,但大多数恶性肿瘤都显示某种程度的分化。
一、肿瘤组织结构的异型性
良性肿瘤瘤细胞的异型性不明显,一般都与其发源组织相似。因此,这种肿瘤的诊断有赖于其组织结构的异型性。例如纤维瘤的细胞和正常纤维细胞很相似,只是其排列与正常纤维组织不同,呈编织状。恶性肿瘤的组织结构异型性明显,瘤细胞排列更为紊乱,失去正常的排列结构层次。例如,从纤维组织发生的恶性肿瘤—纤维肉瘤,瘤细胞很多,胶原纤维很少,排列很紊乱,与正常纤维组织的结构相差较远;从腺上皮发生的恶性肿瘤—腺癌,其腺体的大小和形状十分不规则,排列也较乱,腺上皮细胞排列紧密重叠或呈多层,并可有乳头状增生。
二、肿瘤细胞的异型性
良性肿瘤细胞的异型性小,一般与其发源的正常细胞相似。恶性肿瘤细胞常具有高度的特异性,表现为以下特点:
1.瘤细胞的多形性即瘤细胞形态及大小不一致。恶性肿瘤细胞一般比正常细胞大,各个瘤细胞的大小和形态又很不一致,有时出现瘤巨细胞(图7-2)。但少数分化很差的肿瘤,其瘤细胞较正常细胞小、圆形,大小也比较一致。
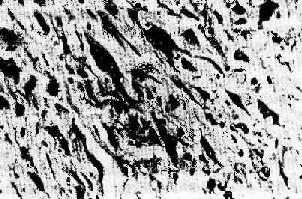
图7-2 横纹肌肉瘤
瘤细胞大小、形状不一,并有瘤巨细胞出现
2.即瘤细胞核的大小、形状及染色不一致。细胞核的体积增大(核肥大),胞核与细胞浆的比例比正常增大(正常为1:4~6,恶性肿瘤细胞则接近1:1),核大小及形状不一,并可出现巨核、双核、多核或奇异形的核,核染色深(由于核内DAN增多),染色质呈粗颗粒状,分布不均匀,常堆积在核膜下,使核膜显得增厚。核仁肥大,数目也常增多(可达3~5个)。核分裂像常增多,特别是出现不对称性、多极性及顿挫性等病理性核分裂像时,对于诊断恶性肿瘤具有重要的意义。恶性肿瘤细胞的核异常改变多与染色体呈多倍体(polyploidy)或非整倍体(aneuploidy)有关。
3.胞浆的改变由于胞浆内核蛋白体增多,胞浆呈嗜碱性。并可因为瘤细胞产生的异常分泌物或代谢产物而具有不同特点,如激素、粘液、糖原、脂质、角质和色素等。
上述瘤细胞的形态,特别是胞核的多形性常为恶性肿瘤的重要特征,在区别良恶性肿瘤上有重要意义。
4.肿瘤细胞超微结构的异型性由于肿瘤类型不同,瘤细胞的超微结构亦有差异。一般说来,良性肿瘤细胞的超微结构基本上与其起源细胞相似。恶性肿瘤细胞根据分化的高低而表现出不同程度的异型性。总的说来,恶性肿瘤细胞的核通常绝对或相对明显增大,核膜可有内陷或外凸,从而使核形不规则甚至形成怪形核。核仁体积增大,数目增多,形状亦可不规则,位置往往靠边。染色质可表现为异染色质增加,凝集成块状散在或(和)边集在核膜下。此即光镜下所见的深染核和核膜增厚;也可表现为常染色质为主,即光镜下所见染色质呈细颗粒状的淡染核。胞浆内的细胞器常有数目减少、发育不良或形态异常,这在分化低的恶性肿瘤细胞更为明显。胞浆内主要见游离的核蛋白体,而其他细胞器大为减少。说明瘤细胞主要合成供自身生长的蛋白质,而丧失其分化的功能,这与瘤细胞生长迅速和恶性程度高有关。溶酶体在侵袭性强的瘤细胞中常有增多,它可释放出大量水解酶,为瘤细胞的浸润开路。细胞间连接常有减少,这在分化低的癌更为突出。可引起细胞间粘着松散,有利于细胞浸润生长。有些在一般情况下在细胞膜表面无微绒毛的瘤细胞也可出现一些不规则微绒毛,这有利于营养物质的吸收,但有碍于细胞间的紧密接触,从而减弱接触抑制,有利于细胞增殖和浸润。瘤细胞的排列结构在分化低的肿瘤也可丧失其原具有的特征改变,例如腺癌细胞可失掉形成腺体的特征而呈弥散排列。因此,恶性肿瘤的超微结构异型性的改变,除了瘤细胞本身的改变外,还可有排列结构的改变。
第三节 肿瘤细胞的代谢特点
肿瘤组织比正常组织代谢旺盛,尤以恶性肿瘤理更为明显。其代谢特点与正常组织相比并无质的差别,但在一定程度上反映了瘤细胞分化不成熟和生长旺盛。
一、核酸代谢
肿瘤组织合成DNA和RNA的聚合酶活性均较正常组织高,与此相应,核酸分解过程明显降低,故DNA和RNA的含量在恶性肿瘤细胞均明显增高。DNA与细胞的分裂和繁殖有关,RNA与细胞的蛋白质合成及生长有关。因此,核酸增多是肿瘤迅速生长的物质基础。
二、蛋白质代谢
肿瘤组织的蛋白质合成及分解代谢都增强,但合成代谢超过分解代谢,甚至可夺取正常组织的蛋白质分解产物,合成肿瘤本身所需要的蛋白质,结果可使机体处于严重消耗的恶病质(cachexia)状态。肿瘤的分解代谢表现为蛋白质分解为氨基酸的过程增强,而氨基酸的分解代谢则减弱,可使氨基酸重新用于蛋白质合成。这可能与肿瘤生长旺盛有关。肿瘤组织还可以合成肿瘤蛋白,作为肿瘤特异抗原或肿瘤相关抗原,引起机体的免疫反应。有的肿瘤蛋白与胚胎组织有共同的抗原性,亦称为肿瘤胚胎性抗原。例如肝细胞癌能合成胎儿肝细胞所产生的甲种胎儿蛋白(AFP),此外,卵巢、睾丸含有卵黄囊结构的生殖细胞肿瘤患者血中AFP也有升高;内胚层组织发生的一些恶性肿瘤如结肠瘤、直肠癌等可产生癌胚抗原(CEA);胃癌可产生胎儿硫糖蛋白等。虽然这些抗原并无肿瘤特异性,也不是肿瘤所专有,但检查这些抗原,并结合其他改变可帮助诊断相应的肿瘤和判断治疗后有无复发。
三、酶系统
肿瘤组织酶活性的改变是复杂的。除了一般在恶性肿瘤组织内氧化酶(如细胞色素氧化酶及琥珀酸脱氢酶)减少和蛋白分解酶增加外,其他酶的改变在各种肿瘤间很少是共同的,而且与正常组织比较只是含量的改变或活性的改变,并非是质的改变。例如前列腺癌的癌组织中酸性磷酸酶明显增加,在前列腺癌伴有广泛骨转移时,患者血清中的酸性磷酸酶也明显增加;骨肉瘤及肝癌时碱性磷酸酶增加,这不但见于肿瘤组织中,还可见于病人的血清中。这些均有助临床诊断。
各种不同组织来源的恶性肿瘤特别是细胞分化原始幼稚者,其酶变化特点主要表现为某些特殊功能的酶接近或完全消失,并因而导致酶谱的一致性。例如分化差的肝癌组织中有关尿素合成的特殊酶系几乎完全消失。其酶谱乃因而趋向与其他癌组织的酶谱一致,与胚胎细胞的酶谱相似。有时还可出现通常所没有的酶。
四、糖代谢
大多数正常组织在有氧时通过糖的有氧分解获取能量,只有在缺氧时才进行无氧糖酵解。肿瘤组织则即使在氧供应充分的条件下也主要是以无氧糖酵解获取能量。这可能是由于癌细胞线粒体的功能障碍所致,或者与瘤细胞的酶谱变化,特别是与三个糖酵解关键酶(己糖激酶、磷酸果糖激酶和丙酮酸激酶)活性增加和同工酶谱的改变,以及糖异生关键酶活性降低有关。糖酵解的许多中间产物被瘤细胞利用合成蛋白质、核酸及脂类,从而为瘤细胞本身的生长和增生提供了必需的物质基础。
第四节 肿瘤的生长与扩散
具有局部浸润和远处转移能力是恶性肿瘤细胞特有的性质。并且是恶性肿瘤威胁病人健康与生命的主要原因。因此对肿瘤生长与扩散的研究已成为肿瘤病理学的重要内容。
一、肿瘤生长的生物学
典型的恶性肿瘤的自然生长史可以分成几个阶段:细胞的恶性转化→转化细胞的克隆性增生→局部浸润→远处转移。在此过程中,恶性转化细胞的内在特点(如肿瘤细胞倍增时间)和宿主对肿瘤细胞或其产物的反应(如肿瘤血管形成)共同影响肿瘤的生长与演进。
1.肿瘤生长动力学肿瘤的生长速度取决于三个因素:
(1)肿瘤细胞倍增时间:恶性转化细胞的生长周期与正常细胞一样,分为G0、G1、S、G2和M期。多数恶性肿瘤细胞的培增时间并不是想象的那样比正常细胞更快,而是与正常细胞相似或者长于正常细胞。
(2)生长分数(growth fraction);生长分数指肿瘤细胞群体中处于复制阶段(S+G2期)的细胞的比例。在细胞恶性转化的初期,绝大多数的细胞处于复制期,所以生长分数很高。但是随着肿瘤的持续生长,不断有瘤细胞发生分化,离开复制阶段的细胞越来越多,使得大多数肿瘤细胞处于G0期。即使是生长迅速的肿瘤其生长分数也只在20%左右。
(3)瘤细胞的生成与丢失:肿瘤的进行性生长及其生长速度决定于其细胞的生成大于丢失的程度。由于营养供应不足,坏死脱落以及机体抗肿瘤反应等因素的影响,在肿瘤生长过程中,有相当一部分瘤细胞失去生命力。肿瘤细胞的生成与丢失的程度共同影响着肿瘤的生长。在生长分数相对较高的肿瘤,瘤细胞的生成远大于丢失,因此其生长速度比那些细胞生成稍超过丢失的肿瘤要快得多。
肿瘤的细胞动力学概念在肿瘤的化学治疗上有重要的意义。目前几乎所有的化学抗癌药物均针对处于复制期的细胞。因此高生长分数的肿瘤(如高恶性的淋巴瘤)对于化疗特别敏感;常见的实体瘤(如结肠癌)生长分数低,故对治疗出现相对耐药性。临床治疗这些肿瘤的战略是先用放射或手术治疗将肿瘤缩小,使残存的瘤细胞从G0期进入复制期后再用化疗。
2.肿瘤的血管形成临床与动物实验都证明,如果没有新生的血管供应营养,肿瘤在达到1~2mm的直径或厚度后,即107个细胞左右将不再增大。因此诱导血管的生成能力是恶性肿瘤能生长、浸润与转移的前提之一。现已发现,由肿瘤细胞本身和浸润到肿瘤组织内及其周围的炎细胞(主要是巨噬细胞)能产生一类血管生成因子(angiogenesis factor),如纤维母细胞生长因子(fibroblastic growth factor,FGF)、血小板衍生的内皮细胞生长因子(platelet-derived endothelial cell growth factor,PD-ECGF)、转化生长因子α(transforming growth factor-α,TGF-α)、肿瘤坏死因子α(TNF-α)、血管内皮细胞生长因子(vascular endothelial growth factor,VEGF)等。其中最具有特征性血管生成作用的是由肿瘤细胞产生的FGF,它能通过其受体与相应的靶细胞结合。有增加内皮细胞的化学趋向性、促进血管内皮细胞分裂、毛细血管出芽生长、诱导蛋白溶解酶生成和有利于内皮细胞芽穿透基质等功能。此外,巨噬细胞产生的TNF-α也有促进内皮细胞分裂和刺激其游走的作用。新生的毛细血管既为肿瘤的生长提供了营养,又为肿瘤的转移准备了条件。因此对抑制肿瘤血管生成的研究是当前的研究热点之一。
3.肿瘤的演进与异质化恶性肿瘤在生长过程中变得越来越富有侵袭性的现象称为肿瘤的演进(progression),包括生长加快、浸润周围组织和远处转移等。这些生物学现象的出现与肿瘤细胞的不同亚克隆在侵袭能力、生长速度、对激素的反应、对抗癌药的敏感性等方面的差异—肿瘤的异质性(heterogeneity)有关。产生这种现象的原因是在肿瘤的生长过程中,可能有附加的基因突变(详见第十五节)作用于不同的瘤细胞,使得瘤细胞的亚克隆获得不同的特性。例如需要较多生长因子的亚克隆可因生长因子缺乏而不能生长,而有些需要较少生长因子的亚克隆在此时即可生长;机体的抗肿瘤反应可杀死那些具有较高的抗原性的亚克隆,而抗原性低的亚克隆则可以躲过机体的免疫监视。由于这些选择,肿瘤在生长过程中能保留那些适应存活、生长、浸润与转移的亚克隆。这就是肿瘤的异质化。
二、肿瘤的生长方式和扩散
1.肿瘤的生长速度各种肿瘤的生长速度有极大的差异,主要决定于肿瘤细胞的分化成熟程度。一般来讲,成熟程度高、分化好的良性肿瘤生长较缓慢,几年甚至几十年。如果其生长速度突然加快,就要考虑发生恶性转变的可能。成熟程度低、分化差的恶性肿瘤生长较快,短期内即可形成明显的肿块,并且由于血管形成及营养供应相对不足,易发生坏死,出血等继发改变。
2.肿瘤的生长方式肿瘤可以呈膨胀性、外生性和浸润性生长:
(1)膨胀性生长:这是大多数良性肿瘤所表现的生长方式。由于这种瘤细胞生长缓慢,不侵袭周围正常组织,随着肿瘤体积的逐渐增大,有如逐渐膨胀的气球,向四周组织推挤。因此肿瘤往往呈结节状,周围常有完整的包膜,与周围组织分界清楚(图7-3)。位于皮下者临床触诊时可以推动,容易手术摘除,摘除后也不易复发。虽这种生长方式的肿瘤对局部器官、组织的影响主要为挤压或阻塞,一般均不明显破坏器官的结构和功能。
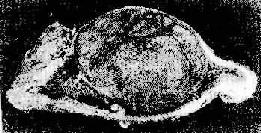
图7-3 良性肿瘤的膨胀性生长(肾上腺皮质腺瘤)
肿瘤呈卵圆形,有包膜,与周围肾上腺组织分界清楚
(2)外生性生长:发生在体表、体腔表面或管道器官(如消化道,泌尿生殖道等)表面的肿瘤,常向表面生长,形成突起的乳头状、息肉状、蕈状或菜花状的肿物。这种生长方式称为外生性生长。良性肿瘤和恶性肿瘤都可呈外生性生长。但恶性肿瘤在外生性生长的同时,其基底部往往也呈浸润性生长,又由于其生长迅速,血液供应不足,这种外生性肿物容易发生坏死脱落而形成底部高低不平、边缘隆起的癌性溃疡。
(3)浸润性生长:为大多数恶性肿瘤的生长方式。瘤细胞分裂增生,侵入周围组织间隙、淋巴管或血管内,如树根之长入泥土,浸润并破坏周围组织。因而此类肿瘤没有包膜,与邻近的正常组织紧密连接在一起而无明显界限(图7-4)。临床触诊时,肿瘤固定不活动。手术切除这种肿瘤时,切除范围比肉眼所见肿瘤范围为大,因为这些部位也可能有肿瘤细胞的浸润。

图7-4 恶性肿瘤的浸润性生长(食管鳞状细胞癌)
癌细胞形成大小、形状不一的细胞巢,浸润于管壁组织(包括肌层)间隙内
3.肿瘤的扩散具有浸润性生长的恶性肿瘤,不仅可以在原发部位继续生长、蔓延(直接蔓延)而且还可以通过多种途径扩散至身体其他部位(转移)。
(1)直接蔓延:随着肿瘤的不断长大,瘤细胞常常连续不断地沿着组织间隙、淋巴管、血管或神经束衣侵入并破坏邻近正常器官或组织,并继续生长,称为直接蔓延。例如晚期子宫颈癌可蔓延至直肠和膀胱;晚期乳腺癌可穿过胸肌和胸腔甚至达肺。
(2)转移(metastasis):瘤细胞从原发部位侵入淋巴管、血管或体腔,被带到他处而继续生长,形成与原发瘤同样类型的肿瘤,这个过程称为转移。所形成的肿瘤称为转移瘤或继发瘤。良性肿瘤不转移,只有恶性肿瘤才可能发生转移。常见的转移途径有以下几种:
1)淋巴道转移:瘤细胞侵入淋巴管(图7-5)后,随淋巴流首先到达局部淋巴结。例如乳腺外上象限发生的乳腺癌首先到达同侧腋窝淋巴结;肺癌首先到达肺门淋巴结。瘤细胞到达局部淋巴结后,先聚集于边缘窦,以后生长繁殖而累及整个淋巴结(图7-6),使淋巴结肿大,质地变硬,切面常呈灰白色。有时有转移的淋巴结由于瘤组织侵出被膜而互相融合成团块。局部淋巴结发生转移后,可继续转移至下一站的其他淋巴结,最后可经胸导管进入血流再继发血道转移。
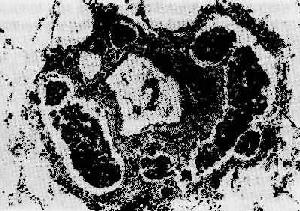
图7-5 肿瘤的淋巴道转移
肺内血管周围淋巴管扩张,充满瘤细胞团(瘤细胞栓子)
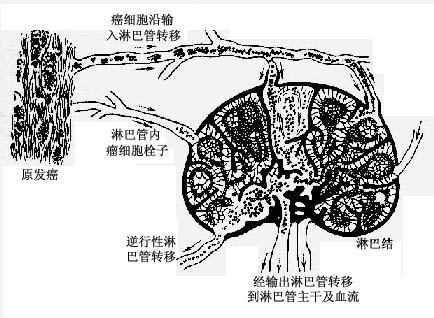
图7-6癌的淋巴道转移模式图
—→淋巴流向 ……→癌细胞流向
2)血道转移:瘤细胞侵入血管后可随血流到达远隔器官继续生长,形成转移瘤。由于动脉壁较厚,同时管内压力较高,故瘤细胞多经小静脉入血。少数亦可经过淋巴管入血。血道转移的运行途径与血栓栓塞过程相同,即侵入体循环静脉的肿瘤细胞经右心到肺,在肺内形成转移瘤,例如骨肉瘤等的肺转移;侵入门静脉系统的肿瘤细胞,首先形成肝内转移,例如胃、肠癌的肝转移等;侵入肺静脉的肿瘤细胞或肺内转移瘤通过肺毛细血管而进入肺静脉的瘤细胞,可经左心随主动脉血流到达全身各器官,常见转移脑、骨、肾及肾上腺等处。因此,这些器官的转移瘤常发生在肺已有转移之后。此外,侵入胸、腰、骨盆静脉的肿瘤细胞,也可以通过吻合支进入脊椎静脉丛(Batson脊椎静脉系统),例如前列腺癌就可通过此途径转移到脊椎,进而转移到脑,这时可不伴有肺的转移。
血道转移虽然可见于许多器官,但最常见的是肺,其次是肝。故临床上判断有无血道转移,以确定患者的临床分期和治疗方案时,作肺部的X线检查及肝的超声等影像学探查是非常必要的。转移瘤在形态上的特点是边界清楚并常为多个散在分布的结节,且多接近器官的表面(图7-7)。位于器官表面的转移瘤,由于瘤结节中央出血、坏死而下陷,可形成“癌脐”。
3)种植性转移:体腔内器官的肿瘤蔓延而于器官表面时,瘤细胞可以脱落并象播种一样,种植在体腔和体腔内各器官的表面,形成多数的转移瘤。这种转移的方式称为种植性转移或播种。种植性转移常见于腹腔器官的癌瘤。如胃癌破坏胃壁侵及浆膜后,可种植到大网膜、腹膜、腹腔内器官表面甚至卵巢等处。肺癌也常在胸腔内形成广泛的种植性转移。浆膜腔的种植性转移每伴有浆液性血性积液。这是由于浆膜下淋巴管或毛细血管被癌栓阻塞,或浆膜受癌的刺激,使其内毛细血管的通透性增加而致渗出增多,并由于血管被癌细胞破坏而引起出血这故。抽吸积液作细胞学检查常可查见癌细胞。脑部的恶性肿瘤,如小脑的髓母细胞瘤(medulloblastoma)亦可经脑脊液转移到脑的其他部位或脊髓,形成种植性转移。值得注意的是,手术也可能造成种植转移,应注意尽量避免。

图7-7 肺内的血道转移性癌
肺的切面上可见多数大小不等的图形癌结节,边界清楚
4.恶性肿瘤的浸润和转移机制
(1)局部浸润:恶性肿瘤局部浸润的机制目前尚未十分明了,但已知是一个由一系列步骤组成的复杂过程。前面提到的浸润能力强的瘤细胞亚克隆的出现和肿瘤血管形成对此都起着重要的作用。局部浸润发生时,由细胞粘附分子(cell adhesion molecules)介导的肿瘤细胞彼此之间的粘附力减弱和瘤细胞与基质的附着力增加是近年来关于侵润机制研究的重点。已有资料显示,在动物高浸润性的肿瘤细胞株,其瘤细胞表面的一种粘附分子——上皮粘连素(E—cadherin)的表达减少,而如将编码上皮粘连素DNA插入到瘤细胞基因组中,则可使其丧失转移和浸润能力。瘤细胞彼此分散才能侵入细胞外基质(extracellular matrix,ECM)。细胞外基质在机体内分隔上皮组织和结缔组织,包括基底膜和间质性结缔组织,主要是由胶原、糖蛋白和蛋白多糖组成。正常上皮细胞与基质的结合是通过存在于上皮细胞膜表面的整合素(integrin)的粘附分子与存在于基质中的其配体的结合来实现的。
癌细胞的ECM主要成成分——基底膜的侵袭是一主动过程,可分为三个步骤:①癌细胞附着于基底膜(attachment):正常上皮细胞具有的一种整合素——层粘连蛋白(laminin,LN)的受体,只分布在细胞的基底面,能与基底膜的LN分子结合而使上皮细胞附着。而癌细胞则有更多的LN受体,分布于癌细胞的整个表面,使癌细胞更容易与基底膜粘附。例如已发现,人的侵润性乳腺癌细胞与LN的结合能力为正常或良性乳腺上皮细胞的50倍,这类病人发生淋巴结转移的机会大大高于LN受体较少的乳腺癌病人。纤维粘连蛋白(fibronectin,FN)也是基底膜的成分,可与上皮细胞表面的一种整合素——FN受体结合而使细胞附着于基底膜。癌细胞FN受体表达增加和其侵袭性之间也存在于与LN类似的关系;②细胞外基质的降解(degradation);在癌细胞与基底膜紧密接触4~8小时后,ECM的成分,如LN、FN、蛋白多糖和胶原纤维(Ⅳ型)可被癌细胞直接分泌的蛋白溶解酶(包括Ⅳ型胶原酶、尿激酶型胞浆素原活化物、组织蛋白酶D等)溶解,使基底膜产生局部的缺损。癌细胞也可诱导宿主细胞(如纤维母细胞)产生蛋白酶,使ECM溶解。Ⅳ型胶原酶是一种金属蛋白酶,能分解上皮和血管基底膜的Ⅳ型胶原纤维,已有报告指出在乳腺癌和胃癌细胞有这种酶的过度表达。动物实验还发现Ⅳ型胶原酶的抑制剂可以大大减少转移的发生;③癌细胞的移出(migration);癌细胞通过被溶解的基底膜缺损处游出是借助于自身的阿米巴运动。近来发现肿瘤细胞的衍生的细胞激肽,如自分泌移动因子(autocrine motility factor)可介导瘤细胞的移动。基质成分(如胶原、LN)的降解产物和某些生长因子(如胰岛素样生长因子Ⅰ和Ⅱ)对癌细胞有化学趋向性。癌细胞穿过基底膜后,重复上述步骤进一步溶解间质性的结缔组织,在间质中移动。到达血管壁时,可以以同样方式穿过血管的基底膜进入血管(图7-8)。
(2)血行播散:进入血管的癌细胞形成新的转移灶的可能性小于千分之一,因单个癌细胞进入血管后绝大多数为机体的免疫细胞消灭。但被血小板凝集成团的癌细胞形成的瘤栓则不易被消灭,并可与形成栓塞处的血管内皮细胞粘附,然后以前述机制穿过血管内皮和基底膜,形成新的转移灶(图7-8)。由于肿瘤的异质化而选择出的高侵袭性的瘤细胞亚克隆,尤其容易形成广泛的血行播散。
转移的发生不是随机的。早在1889年Paget在对700多侧乳腺癌的转移进行分析后,就发现有明显的器官倾向性,并提出有名的“种子和土壤”学说。血行转移的位置和器官分布,在某些肿瘤具有特殊的亲和性,如肺癌易转移到肾上腺和脑;甲状腺癌、肾癌和前列腺癌易转移到骨;乳腺癌常转移到肺、肝、骨、卵巢和肾上腺等。产生这种现象的原因还不清楚,可能是由于这些器官的血管内皮上有能与进入血循环的癌细胞表面的粘附分子特异性结合的配体(如血管细胞粘附分子),或者由于靶器官能够释放某些吸引癌细胞的化学吸引物质(如胰岛素样生长因子Ⅰ和Ⅱ)。此外,转移瘤在某些组织或器官中不易形成,也可能与这些器官或组织的环境不适合肿瘤的生长有关。如脾虽然血液循环丰富但转移癌少见,可能是因为脾是免疫器官;横纹肌转移瘤很少,可能是因为肌肉经常收缩使瘤细胞不易停留或肌肉内乳酸含量过高,不利于肿瘤生长。
(3)肿瘤转移的分子遗传学:目前尚发现一个单独的转移基因,但已发现一种肿瘤抑制基因——nm23的表达水平与肿瘤的侵袭和转移能力之间存在有意义的关系。在小鼠模型中,nm23的表达高者具有低转移性;nm23表达低10倍者伴有高转移。人类的nm23基因定位于第17位号染色体。在侵袭性强的肿瘤中nm23基因丢失。临床上对人乳腺癌的观察发现,淋巴结转移少于三个者,nm23蛋白表达水平高;而有广泛转移者nm23蛋白表达的水平一般均低。如能将nm23蛋白作为标记物来预测转移并且在治疗上用于抑制转移,将是肿瘤治疗的一个突破。现此问题正在积极研究中。
三、肿瘤的分级与分期
肿瘤的分级(grading)和分期(staging)一般都用于恶性肿瘤。恶性肿瘤是根据其分化程度的高低、异型性的大小及核分裂像的多少来确定恶性程度的级别。近年来较多的人倾向于用简明的、较易掌握的三级分级法,即Ⅰ级为分化良好的,属低度恶性;Ⅱ级为分化中等的,属中度恶性;Ⅲ级为分化低的,属高度恶性。这种分级法虽有其优点,对临床治疗和判断预后也有一定意义。但缺乏定量标准,也不能排除主观因素的影响。因此,如何建立精确的分级标准还待进一步研究。
肿瘤的分期目前有不同的方案,其主要原则是根据原发肿瘤的大小、浸润的深度、范围以及是否累及邻近器官、有无局部和远处淋巴结的转移、有无血源性或其他远处转移等来确定肿瘤发展的程期或早晚。
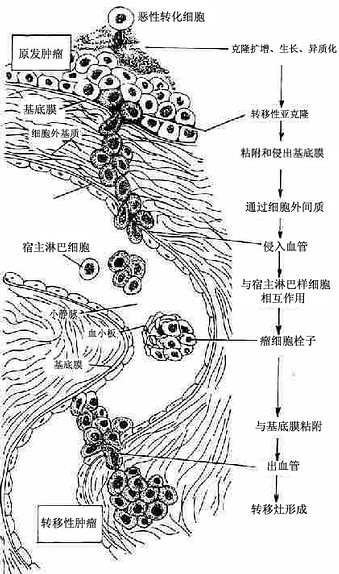
图7-8 恶性肿瘤浸润和转移机制
恶性肿瘤细胞侵出基底膜进入细胞外间质,侵入血管并形成新的转移灶(采自Basic Pathology)
肿瘤的分级和分期对临床上制定治疗方案和估计预后有参考价值,特别是肿瘤的分期更为重要,但是必须结合各种恶性肿瘤的生物学特性以及病人的全身情况等综合考虑。
第五节 肿瘤对机体的影响
肿瘤因其良恶性的不同,对机体的影响也有所不同。
良性肿瘤:因其分化较成熟,生长缓慢,停留于局部,不浸润,不转移,故一般对机体的影响相对较小,主要表现为局部压迫和阻塞症状。其影响的发生主要与其发生部位和继发变化有关。如体表良性瘤除少数可发生的局部症状外,一般对机体无重要影响;但若发生在腔道或重要器官,也可引起较为严重的后果,如消化道良性肿瘤(如突入肠腔的平滑肌瘤),有时引起肠梗阻或肠套叠;颅内的良性瘤(如脑膜瘤、星形胶质细胞瘤)可压迫脑组织阻塞脑室系统而引起颅内压升高和相应的神经系统症状。良性肿瘤有时可发生继发性改变,亦可对机体带来程度不同的影响,如肠的腺瘤性息肉、膀胱的乳头状瘤等表面可发生溃疡而引起出血和感染。此外,内分泌腺的良性肿瘤则常因能引起某种激素分泌过多而产生全身性影响,如垂体前叶的嗜酸性腺瘤(acidophil adenoma)可引起巨人症(gigantism)或肢端肥大症(acromegaly);胰岛细胞瘤分泌过多的胰岛素,可引起阵发性血糖过低等。
恶性肿瘤:恶性肿瘤由于分泌不成熟、生长较快,浸润破坏器官的结构和功能,并可发生转移,因而对机体的影响严重。恶性瘤除可引起与上述良性瘤相似的局部压迫和阻塞症状外,发生于消化道者更易并发溃疡、出血、甚致穿孔,导致腹膜炎,后果更为严重。有时肿瘤产物或合并感染可引起发热。肿瘤浸润、压迫局部神经还可引起顽固性疼痛等症状。恶性肿瘤的晚期患者,往往发生恶病质(cachexia),可致患者死亡。恶病质是指机体严重消瘦、无力、贫血和全身衰竭的状态,其发生机制尚未完全阐明,可能由于缺乏食欲,进食减少、出血、感染、发热或因肿瘤组织坏死所产生的毒性产物等引起机体的代谢紊乱所致。此外,恶性肿瘤的迅速生长,消耗机体大量的营养物质,以及由于晚期癌瘤引起的疼痛,影响患者的进食及睡眠等,也是招致恶病质的重要因素。近年来发现巨噬细胞产生的肿瘤坏死因子(TNF)可降低食欲和增加分解代谢,与恶病质的发生有一定关系。
近年来相继发现一些非内分泌腺肿瘤能产生和分泌激素或激素类物质,如促肾上腺皮质激素(ACTH)、甲状旁腺素(PTH)、胰岛素、抗利尿激素(ADH)、人绒毛膜促性腺激素(HCG)、促甲状腺激素(TSH)、生长激素(GH)、降钙素(calcitonin)等十余种,可引起内分泌紊乱的临床症状。此类肿瘤称为异位内分泌肿瘤,其所引起的临床症状称为异位内分泌综合征。此类肿瘤大多为恶性肿瘤,其中以癌为多,如肺癌、胃癌、肝癌、胰腺癌、结肠癌等;也可见于肉瘤,如纤维肉瘤,平滑肌肉瘤、横纹肌肉瘤和未分化肉瘤等。许多分泌异位激素的恶性肿瘤都有产生两种以上激素的特点。关于异位激素产生的机制于今尚无一致的解释,可能与瘤细胞内基因异常表达有关。除上述异位内分泌综合征外,由于肿瘤的产物或异常免疫反应(包括交叉免疫、自身免疫和免疫复合物沉着等)或其他不明原因,还可引起神经、消化、造血、骨关节、肾及皮肤等系统发生一些病变和临床表现。这些表现不是由原发肿瘤或转移灶所在部位直接引起的,而是通过上述途径间接引起,故称为副肿瘤综合征(paraneoplastic syndrome)。有时这些综合征可以表现得非常明显而造成严重后果。认识此种综合征的意义在于它可能是一些隐匿肿瘤的早期表现,可由此而发现早期肿瘤。再者不要误认为这些系统的改变是由肿瘤转移所致,而放弃对肿瘤的治疗。与之相反,如肿瘤治疗有效,这些综合征可减轻或消失。因此有十分重要的临床意义。
第七节 良性肿瘤与恶性肿瘤的区别
综上所述,良性肿瘤和恶性肿瘤在生物学特点上是明显不同的,因而对机体的影响也不同。良性肿瘤一般对机体影响小,易于治疗,疗效好;恶性肿瘤危害较大,治疗措施复杂,疗效还不够理想。如果把恶性肿瘤误诊为良性肿瘤,就会延误治疗或治疗不彻底,造成复发、转移。相反,如把良性肿瘤误诊为恶性肿瘤,也必然要进行一些不必要、不恰当的治疗,使患者遭受不应有的痛苦、损害和精神负担。因此,区别良性肿瘤与恶性肿瘤,对于正确的诊断和治疗具有重要的实际意义。现将良性肿瘤与恶性肿瘤的区别列于表7-1。
表7-1 良性肿瘤与恶性肿瘤的区别
| 良性肿瘤 | 恶性肿瘤 | |
| 组织分化程度 | 分化好,异型性小,与原有组织的形态相似 | 分化不好,异型性大,与原有组织的形态差别大 |
| 核分裂 | 无或稀少,不见病理核分裂像 | 多见,并可见病理核分裂像 |
| 生长速度 | 缓慢 | 较快 |
| 生长方式 | 膨胀性和外生性生长,前者常有包膜形成,与周围组织一般分界清楚,故通常可推动 | 浸润性和外生性生长,前者无包膜,一般与周围组织分界不清楚,通常不能推动,后者每伴有浸润性生长 |
| 继发改变 | 很少性发生坏死、出血 | 常发生出血、坏死、溃疡形成等 |
| 转移 | 不转移 | 常有转移 |
| 复发 | 手术后很少复发 | 手术等治疗后较多复发 |
| 对机体影响 | 较小,主要为局部压迫或阻塞作用。如发生在重要器官也可引起严重后果 | 较大,除压迫、阻塞外,还可以破坏原发处和转移处的组织,引起坏死出血合并感染,甚至造成恶病质 |
必须指出,良性肿瘤与恶性肿瘤间有时并无绝对界限,有些肿瘤其表现可以介乎两者之间,称为交界性肿瘤(如卵巢交界性浆液性乳头状囊腺瘤和粘液性囊腺瘤)。此类肿瘤有恶变倾向,在一定的条件下可逐渐向恶性发展。在恶性肿瘤中,其恶性程度亦各不相同,有的较早发生转移,如鼻咽癌;有的转移晚,如子宫体腺癌;有的则很少发生转移。此外,肿瘤的良恶性也并非一成不变,有些良性肿瘤如不及时治疗,有时可转变为恶性肿瘤,称为恶性变(malignant change),如结肠腺瘤性息肉可恶变为腺癌。而个别的恶性肿瘤如黑色素瘤,有时由于机体免疫力加强等原因,可以停止生长甚至完全自然消退。又如见于少年儿童的神经母细胞瘤(neuroblastoma)的瘤细胞有时能发育成为成熟的神经细胞,有时甚至转移灶的瘤细胞也能继续分化成熟,使肿瘤停止生长而自愈。但这种情况毕竟罕见,绝大多数恶性肿瘤能否逆转为良性,是目前肿瘤研究的重要课题之一。
第六节 肿瘤的命名与分类
一、肿瘤的命名原则
人体任何部位、任何组织、任何器官几乎都可发生肿瘤,因此肿瘤的种类繁多,命名也复杂。一般根据其组织发生即组织来源来命名。良性肿瘤在其来源组织名称后加“瘤”字,例如来源于纤维结缔组织的良性瘤称为纤维瘤(fibroma),来源于腺上皮的良性瘤称为腺瘤(adenoma)等。有时还结合肿瘤的形态特点命名,如腺瘤呈乳头状生长并有囊腔形成者称为乳状头状囊腺瘤。恶性肿瘤一般亦根据其组织来源来命名。来源于上皮组织的统称为癌(carcinoma),命名时在其来源组织名称之后加“癌”字,如来源于鳞状上皮的恶性肿瘤称为鳞状细胞癌,来源于腺上皮呈腺样结构的恶性肿瘤称为腺癌等。从间叶组织(包括纤维结缔组织、脂肪、肌肉、脉管、骨、软骨组织等)发生的恶性肿瘤统称为肉瘤(sarcoma),其命名方式是在来源组织名称之后加“肉瘤”,例如纤维肉瘤、横纹肌肉瘤、骨肉瘤等。恶性肿瘤的外形具有一定的特点时,则又结合形态特点而命名,如形成乳头状及囊状结构的腺癌,则称为乳头状囊腺癌。如一个肿瘤中既有癌的结构又有肉瘤的结构,则称为癌肉瘤(carcinosarcoma)。
在病理学上,癌是指上皮组织来源的恶性肿瘤,但一般人所说的“癌症”(cancer),习惯上常泛指所有恶性肿瘤。
有少数恶性肿瘤不按上述原则命名,如有些来源于幼稚组织及神经组织的恶性肿瘤称为母细胞瘤,如神经母细胞瘤、髓母细胞瘤、肾母细胞瘤等;有些恶性肿瘤成分复杂或由于习惯沿袭,则在肿瘤的名称前加“恶性”,如恶性畸胞瘤、恶性淋巴瘤、恶性黑色素瘤等。有些恶性肿瘤冠以人名、如尤文(Ewing)瘤、何杰金(Hodgkin)病;或按肿瘤细胞的形态命名,如骨的巨细胞瘤、肺的燕表细胞癌。至于白血病、精原细胞癌则是少数采用习惯名称的恶性肿瘤,虽称为“瘤”或“病”,实际上都是恶性肿瘤。
二、肿瘤的分类
肿瘤的分类通常是以其组织发生(即来源于何种组织)为依据,每一类别又分为良性与恶性两大类。兹举例如下(表7-2)。
表7-2 肿瘤分类举例
| 组织来源 | 良性肿瘤 | 恶性肿瘤 | 好发部位 |
| 一、上皮组织 | |||
| 鳞状上皮 | 乳头状瘤 | 鳞状细胞癌 | 乳头状瘤见于皮肤、鼻、鼻窦、喉等处;鳞癌见于宫颈、皮肤、食管、鼻咽、肺、喉和阴茎等处 |
| 基底细胞 | 基底细胞癌 | 头面部皮肤 | |
| 腺上皮 | 腺瘤 | 腺瘤(各种类型) | 腺瘤多见于皮肤、甲状腺、胃、肠;腺癌见于胃、肠、乳腺、甲状腺等 |
| 粘液性或浆液性囊腺瘤 | 粘液性或浆液性囊腺癌 | 卵巢 | |
| 多形性腺瘤 | 恶性多形性腺瘤 | 涎腺 | |
| 移行上皮 | 乳头状瘤 | 移行上皮癌 | 膀胱、肾盂 |
| 二、间叶组织 | |||
| 纤维结缔组织 | 纤维瘤 | 纤维肉瘤 | 四肢 |
| 纤维组织细胞 | 纤维组织细胞瘤 | 恶性纤维组织细胞瘤 | 四肢 |
| 脂肪组织 | 脂肪瘤 | 脂肪肉瘤 | 前者多见于皮下组织,后者多见于下肢和腹膜后 |
| 平滑组织 | 平滑肌瘤 | 平滑肌肉瘤 | 子宫和胃肠 |
| 横纹肌组织 | 横纹肌瘤 | 横纹肌肉瘤 | 肉瘤多见于头颈、生殖泌尿道及四肢 |
| 血管和淋巴管组织 | 血管瘤、淋巴管瘤 | 血管肉瘤 淋巴管肉瘤 |
皮肤和皮下组织、舌、唇等 |
| 骨组织 | 骨瘤 | 骨肉瘤 | 骨瘤多见于颅骨、长骨;骨肉瘤多见于长骨两端,以膝关节上下尤为多见 |
| 巨细胞瘤 | 恶性巨细胞瘤 | 股骨上下端、胫骨上端、肱骨上端 | |
| 软骨组织 | 软骨瘤 | 软骨肉瘤 | 软骨瘤多见于手足短骨;软骨肉瘤多见于盆骨、肋骨、股骨、肱骨及肩胛骨等 |
| 滑膜组织 | 滑膜瘤 | 滑膜肉瘤 | 膝、踝、肩和肘等关节附近 |
| 间皮 | 间皮瘤 | 恶性间皮瘤 | 胸膜、腹膜 |
| 三、淋巴造血组织 | |||
| 淋巴组织 | 恶性淋巴瘤 | 颈部、纵隔、肠系膜和腹膜后淋巴结 | |
| 造血组织 | 各种白血病 | 淋巴造血组织 | |
| 多发性骨髓瘤 | 椎骨、胸骨、肋骨、颅骨和长骨 | ||
| 四、神经组织 | |||
| 神经衣组织 | 神经纤维瘤 | 神经纤维肉瘤 | 单发性;全身皮神经;多发性:深部神经及内脏也受累 |
| 神经鞘细胞 | 神经鞘瘤 | 恶性神经鞘瘤 | 头、颈、四肢等处神经 |
| 胶质细胞 | 胶质细胞瘤 | 恶性胶质细胞瘤 | 大脑 |
| 原始神经细胞 | 髓母细胞瘤 | 小脑 | |
| 脑膜组织 | 脑膜瘤 | 恶性脑膜瘤 | 脑膜 |
| 交感神经节 | 节细胞神经瘤 | 神经母细胞瘤 | 前者多见于纵隔和腹膜后,后者多见于肾上腺髓质 |
| 五、基他肿瘤 | |||
| 黑色素细胞 | 黑痣 | 恶性黑色素瘤 | 皮肤、粘膜 |
| 胎盘组织 | 葡萄胎 | 绒毛膜上皮癌、恶性葡萄胎 | 子宫 |
| 性索 | 支持细胞、间质细胞瘤 | 恶性支持细胞、间质细胞瘤 | 卵巢、睾丸 |
| 颗粒细胞瘤 | 恶性颗粒细胞瘤 | 卵巢 | |
| 生殖细胞 | 精原细胞瘤 | 睾丸 | |
| 无性细胞瘤 | 卵巢 | ||
| 胚胎性癌 | 睾丸、卵巢 | ||
| 三个胚叶组织 | 畸胎瘤 | 恶性畸胎瘤 | 卵巢、睾丸、纵隔和骶尾部 |
第八节 上皮性肿瘤
从上皮组织(包括覆盖上皮与腺上皮)发生的肿瘤最为常见,其中恶性上皮组织肿瘤(癌)对人类的危害最大,人体的恶性肿瘤大部分来源于上皮组织。
一、良性上皮组织肿瘤
1.乳头状瘤(papilloma)由覆盖上皮发生,向表面呈外生性生长,形成许多手指样或乳头状突起,并可呈菜花状或绒毛状外观。肿瘤的根部常较狭窄成蒂与正常组织相连。镜下,每一乳头由具有血管的分支状结缔组织间质构成其轴心,其表面覆盖的增生上皮因起源部位不同而异,可为鳞状上皮(图7-9)、柱状上皮或移行上皮。在外耳道、阻茎及膀胱和结肠的乳头状瘤较易发生恶变而形成乳头状癌,值得注意。
图7-9 皮肤乳头状瘤
2.腺瘤(adenoma)是由腺上皮发生的良性肿瘤,多见于甲状腺、卵巢、乳腺、涎腺和肠等处。粘膜腺的腺瘤多呈息肉状(图7-10),腺器官内的腺瘤则多呈结节状,且常有包膜,与周围正常组织分界清楚。腺瘤的腺体与其起源腺体不仅在结构上十分相似,而且常具有一定的分泌功能。不同这外仅在于腺瘤的腺体大小、形态较不规则,排列也比较密集。发生于有小叶和导管结构的器官的腺瘤,其小叶结构往往缺如或不明显,亦无导管形成,故不能将其分泌物排出。
图7-10 肠的息肉状腺瘤模式图
根据腺瘤的组成成分或形态特点,又可将其分为囊腺瘤、纤维腺瘤、多形性腺瘤和息肉状腺瘤等类型。
(1)囊腺瘤(cystadenoma):是由于腺瘤组织中的腺体分泌物淤积,腺腔逐渐扩大并互相融合成囊,肉眼上可见大小不等的囊腔,因而得名。囊腺瘤常发生于卵巢,亦偶见于甲状腺及胰腺。卵巢囊腺瘤主要有两种类型:一种为腺上皮向囊腔内呈乳头状生长,并分泌浆液,故称为浆液性乳头状腺瘤(serous papillary cystadenoma);另一种分泌粘液,常为多房性,囊壁多光滑,少有乳头状增生,称为粘液性囊腺瘤(mucinous cystadenoma) (图7-11)。浆液性乳头状囊腺瘤较易发生恶变,转化为浆液性囊腺癌(serous cystadenocarcinoma)。
图7-11 卵巢粘液性囊腺瘤
肿瘤呈多房性囊状,囊腔内含粘液
(2)纤维腺瘤(fibroadenoma):除腺上皮细胞增生形成腺体外,尚同时伴随大量纤维结缔组织增生,共同构成瘤的实质。本瘤为女性乳腺常见的良性肿瘤。
(3)多形性腺瘤(pleomorphic adenoma)由腺组织、粘液样及软骨样组织等多种成分混合组成。常发生于涎腺,特别常见于腮腺,过去增称之为混合瘤。目前一般认为此瘤是由腮腺闰管上皮和肌上皮发生的一种肿瘤。由于分散的肌上皮细胞之间可出现粘液样基质,并可化生为软骨样组织,从而构成多形性特点。本瘤生长缓慢,但切除后较易复发。
(4)息肉状腺瘤(polypous adenoma) 发生于粘膜,呈息肉状,有蒂与粘膜相连,多见于直肠(图7-10),其中表面呈乳头状或绒毛状者恶变率较高。本瘤亦见于结肠、胃等处,结肠多发性腺瘤性息肉常有家族遗传性,不但癌变率很高,并易早期发生癌变。
二、恶性上皮组织肿瘤
由上皮发生的恶性肿瘤称为癌,多见于老年人,是人类最常见的一类恶性肿瘤。癌常以浸润性生长为主,故与周围组织分界不清。发生在皮肤、粘膜表面的癌外观上常呈息肉状、蕈伞状或菜花状,表面常有坏死及溃疡形成;发生在器官内的常为不规则的结节状,呈树根状或蟹足状向周围组织浸润,质地较硬,切面常为灰白色,较干燥。镜下,癌细胞可呈腺状、巢状或条索状排列,与间质分界清楚。亦可在间质内呈弥漫性浸润生长,与间质分界不清。网状纤维染色见癌细胞之间多无网状纤维,而只见于癌巢的周围。癌在早期一般多经淋巴道转移,到晚期才发生血道转移。
癌的常见类型有以下几种:
1.鳞状细胞癌(squamous cell carcinoma)简称鳞癌,常发生在身体原有鳞状上皮覆盖的部位,如皮肤、口腔、唇、子宫颈、阴道、食管、喉、阻茎等处。有些部位如支气管、胆囊、肾盂等处,正常时虽不由鳞状上皮覆盖,但可通过鳞状上皮化生而发生鳞状细胞癌。此癌肉眼上常呈菜花状,也可因癌组织坏死脱落而形成溃疡。癌组织也同时向深层作浸润性生长。镜下,在分化好的鳞状细胞的癌的癌巢中,细胞间还可见到细胞间桥,在癌巢的中央可出现层状的角化物,称为角化珠(keratin pearl)或癌珠(图7-12)。分化较差的鳞状细胞癌无角化珠形成,甚至也无细胞间桥,瘤细胞呈明显的异型性并见较多的核分裂像。
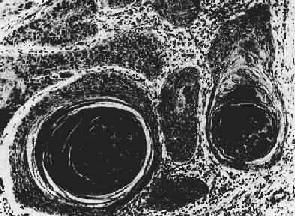
图7-12 鳞状细胞癌
图示典型的癌细胞巢,其中两个癌巢中央形成同心圆状的角化珠
2.基底细胞癌(basal cell carcinoma)多见于老年人面部如眼睑、颊及鼻翼等处,由该处表皮原始上皮芽或基底细胞发生。癌巢主要由浓染的基底细胞样的癌细胞构成,本癌生长缓慢,表面常形成溃疡,并可浸润破坏局部深层组织,但很少发生转移,对放射治疗很敏感,临床上呈低度恶性的经过。
3.移行上皮癌(transitional cell carcinoma)来自膀胱或肾盂等处的移行上皮,常呈乳头状,多发性,可溃破形成溃疡或广泛浸润膀胱壁。镜下,癌细胞似移行上皮,呈多层排列,异型性明显。
4.腺上皮癌是从腺上皮发生的恶性肿瘤。根据其形态结构和分化程度,可分为分化比较好的、具有腺体结构的腺癌和低分化的、形成实体癌巢的实性癌。腺癌分泌粘液较多的则称粘液癌。
(1)腺癌(adenocarcinoma):较多见于胃肠、胆囊、子宫体等。癌细胞形成大小不等,形状不一、排列不规则的腺样结构,细胞常不规则地排列成多层,核大小不一,核分裂像多见(图7-13)。当腺癌伴有大量乳头状结构时称为乳头状腺癌;腺腔高度扩张呈囊状的腺癌称为囊腺癌;伴乳头性生长的囊腺癌称为乳头状囊腺癌。
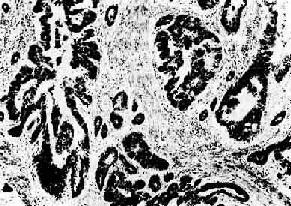
图7-13 腺癌(胃)
癌细胞排列紊乱,多层,形成大小不等、形状不规则的腺样结构,浸润于胃壁组织内
(2)粘液癌(mucoid carcinoma):又称为胶样癌(colloid carcinoma),常见于胃肠。镜下,初时粘液聚积在癌细胞内,将核挤向一侧,使该细胞成印戒状, 故一般称之为印戒细胞(signet-ring cell)。以后粘液堆积在腺腔内,并可由于腺体的崩解而形成粘液池。此时,往往可见于小堆或散在的印戒状癌细胞漂浮其中。肉眼观,癌组织呈灰白色,湿润,半透明如胶冻样,胶样癌因而得名。
(3)实性癌(solid carcinoma):或称单纯癌(carcinoma simplex),属低分化的腺癌,恶性程度较高,多发生于乳腺,少数可发生于胃及甲状腺。癌巢为实体性,无腺腔样结构,癌细胞异型性高,核分裂像多见。有的癌巢小而少,间质结缔组织多,质地硬,称为硬癌(scirrhous carcinoma)(图7-14)。有的则癌巢较大较多,间质结缔组织相对较少,质地如脑髓,称为髓样癌(medullary carcinoma)。
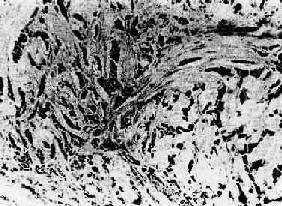
图7-14 乳腺硬癌
癌细胞呈条索状排列,间质丰富,由多量纤维组织构成
三、癌前疾病及癌前病变、非典型性增生及原位癌
正确认识癌前疾病癌前病变、非典型性增生及原位癌是防止肿瘤发生发展及早期诊断肿瘤的重要环节。
1.癌前疾病及癌前病变癌前疾病及癌前病变是指某些统计上具有明显癌变危险的疾病及病变,如不及时治愈即有可能转变为癌。因此,早期发现与及时治愈癌前疾病或癌前病变,对肿瘤的预防具有重要的实际意义。常见的癌前疾病及病变有以下几种:
(1)粘膜白斑:常发生在食管、口腔、子宫颈及外阴等处粘膜。主要病理改变是粘膜的鳞状上皮过度增生和过度角化,并出现一定的异型性。肉眼上呈白色斑块,故称白斑。如长期不愈就有可能转变为鳞状细胞癌。
(2)慢性子宫颈炎伴宫颈糜烂:是妇女常见的疾患。在慢性子宫颈炎的基础上子宫颈阴道部的鳞状上皮被来自子宫颈管内膜的单层柱状上皮所取代,使该处呈粉红色或鲜红色,好象发生了粘膜上皮的缺损,称为子宫颈糜烂。随后,局部又可被再生的鳞状上皮所替代,称为糜烂愈复。如果上述过程反复进行,则少数病例可变为子宫颈鳞状细胞癌。
(3)纤维囊性乳腺病:本病由内分泌失调引起,常见于40岁左右的妇女,主要表现为乳腺小叶导管和腺泡上皮细胞的增生、大汗腺化生及导管囊性扩张,间质纤维组织也有增生。伴有导管内乳头状增生者较易发生癌变。
(4)结肠、直肠的腺瘤性息肉:较为常见,可以单发或多发,均可发生癌变。多发性者常有家族史,更易发生癌变。
(5)慢性萎缩性胃炎及胃溃疡:慢性萎缩性胃炎时,胃粘膜腺体可有肠上皮化生,这种肠上皮化生与胃癌的发生有一定关系,如久治不愈可发生癌变。慢性胃溃疡时溃疡边缘的粘膜因受刺激而不断增生,过去认为有5%可能转变为癌,现在认为其癌变率大约为1%。
(6)慢性溃疡性结肠炎:在溃疡反复和粘膜增生的基础上可发生结肠腺癌。
(7)皮肤慢性溃疡:经久不愈的皮肤溃疡和瘘管,特别是小腿的慢性溃疡,由于长期慢性刺激,表皮鳞状上皮增生,有的可发生癌变。
但须指出,癌的形成往往经历一个漫长的、逐渐演进的过程,平均为15~20年,而且并非所有癌前疾病和病变都必然转变为癌,这还取决于很多因素。再者也并非所有的癌目前都已发现明确的癌前疾病,这方面的研究在肿瘤的预防上具有重要意义。
2.非典型性增生(dysplasia,atypical hyperplasia)主要指上皮细胞导乎常态的增生,表现为增生的细胞大小不一,形态多样,核大而浓染,核浆比例增大,核分裂可增多但多呈正常核分裂像。细胞排列较乱,极向消失。可发生于皮肤或粘膜表面的被覆上皮,也可发生于腺体上皮。根据其异型性程度和(或)累及范围可分为轻、中、重三级。轻度和中度的非典型性增生(只累及上皮下部的1/3~2/3处),在病因消除后可恢复正常。而重度非典型性增生则累及上皮全层并很难逆转,常转变为癌。上述癌前疾病和病变多通过非典型增生而发生癌变的。
3.原位癌原位癌的一般指粘膜上皮层内或皮肤表皮层内的非典型增生(重度)累及上皮的全层,但尚未侵破基底膜向下浸润生长者。例如子宫颈、食管及皮肤的原位癌。此外,当乳腺小叶腺泡发生癌变而尚未浸润至小叶外者,亦可称为小叶原位癌。原位癌是一种早期癌,因而早期发现和积极治疗,可防止其发展为浸润性癌,从而提高癌瘤的治愈率。
第九节 间叶组织肿瘤
一、良性间叶组织肿瘤
这类肿瘤的分化成熟程度高、其组织结构、细胞形态、硬高及颜色等均与其发源的正常组织相似。肿瘤生长慢,呈膨胀性长长,一般都具有包膜。现分述其中比较常见的类型:
1.纤维瘤(fibroma)瘤组织内的胶原纤维排成束状,互相编织,纤维间含有细长的纤维细胞(图7-15)。外观呈结节状,与周围组织分界明显,有包膜。切面灰白色,可见编织状的条纹,质地韧硬,常见于四肢及躯干的皮下。此瘤生长缓慢,手术摘除后不再复发。
瘤样纤维组织增生:纤维结缔组织呈肿瘤样增生,形成瘤样肿块,又称纤维瘤病(fibromatosis)。它包括的种类繁多,约有十多种。常见的有结节性筋膜炎、增生性筋膜炎、增生性肌炎和带状瘤等。瘤样纤维组织增生与纤维瘤不同之处在于局部呈浸润性生长,没有包膜。除见纤维母细胞外,尚见大量的肌纤维母细胞,有时纤维母细胞增生较活跃,细胞丰富,核肥大甚至具有轻度异型性和少数正常的核分裂像,切除不完全时可以多次复发但不转移。最重要的是不要将其误诊为恶性肿瘤。此种病损根据类型的不同,可见于婴儿、儿童和成人,可发生于任何部位的软组织,但以发生在四肢及腹壁的皮下、筋膜、肌肉等处较为多见。
2.脂肪瘤(lipoma)最常见的部位为背、肩、颈及四肢近端的皮下组织。外观为扁圆形或分叶状,有包膜,质地柔软,切面色淡黄,似正常的脂肪组织。肿瘤大小不一,直径由数厘米至数十厘米不等,常为单发性,亦可为多发性。镜下结构与正常脂肪组织的主要区别在于有包膜。瘤组织分叶大小不规则,并有不均等的纤维组织间隔存在。脂肪瘤一般无明显症状,但也有引起在局部疼痛者,很少恶变,手术易切除。
图7-15 纤维瘤
瘤组织由分化良好的纤维细胞构成,呈编织状排列,有丰富的胶原纤维
3.脉管瘤可分为血管瘤(hemangioma)及淋巴管瘤(lymphangioma)两类。其中以血管瘤最为常见,多为先天性发生,故常见于儿童。血管瘤可以生在任何部位,但以皮肤为多见。一般分为毛细血管瘤(由增生的毛细血管构成)(图7-16)、海绵状血管瘤(由扩张的血窦构成)及混合型血管瘤(即二种改变并存)三种。肉眼上无包摸,呈浸润性生长。在皮肤或粘膜可呈突起的新鲜红肿块,或仅呈暗红或紫红色斑。内脏血管瘤多呈结节状。发生于肢体软组织的弥漫性海绵状血管瘤可引起肢体肥大。血管瘤一般随身体的发育而长大,成年后即停止发展,甚至可以自然消退。
图7-16 毛细血管瘤
瘤组织主由大量增生的毛细血管构成,部分毛细血管腔内可见红细胞
淋巴管瘤由增生的淋巴管构成,内含淋巴液。淋巴管可呈囊性扩大并互相融合,内含大量淋巴液,称为囊状水瘤,此瘤多见于小儿。
4.平滑肌瘤(leiomyoma)最多见于子宫,其次为胃肠。瘤组织由形态比较一致的梭形平滑肌细胞构成。细胞排列成束状,互相编织,核呈长杆状,两端钝圆,同一束内的细胞核有时排列成栅状,核分裂像少见。
5.骨瘤(osteoma)本瘤好发于头面骨及颌骨,也可累及四肢骨,形成局部隆起。镜下见主要由成熟的骨质组成,但失去正常骨质的结构和排列方向。骨瘤发生在颅骨内板者可凸向颅腔,引起颅神经压迫症状;发生于眼眶、鼻窦或颌骨者可引起相应部位压迫症状。
6.软骨瘤(chondroma)自软骨膜发生者,称外生性软骨瘤;发生于手足短骨和四肢长骨等骨干的骨髓腔内者称为内生性软骨瘤。内眼观,前者自骨表面突起,常分叶;后者使骨膨胀,外有薄骨壳。切面呈淡蓝色或银白色,半透明,可有钙化或囊性变,内含粘液。镜下见瘤组织由成熟的透明软骨组成,呈不规则分叶状,每一小叶由疏松的纤维血管间质包绕。肿瘤位于盆骨、胸骨、肋骨、四肢长骨或椎骨时易恶变;发生在指(趾)骨者极少恶变。
二、恶性间叶组织肿瘤
恶性间叶组织肿瘤统称为肉瘤。肉瘤比癌少见,多发生于青少年。内眼观呈结节状或分叶状。由于其发生较快,除浸润生长外,也可挤压周围组织形成假包膜。肉瘤体积常较大,质软,切面多呈灰红色,均质性,湿润,外观多呈鱼肉状,故称为肉瘤。肉瘤易发生出血、坏死、囊性变等继发性改变。镜下,肉瘤细胞大多弥漫排列,不形成细胞巢,与间质分界不清,网状纤维染色可见肉瘤细胞间存在网状纤维。肿瘤间质的结缔组织少,但血管较丰富,故肉瘤多先由血道转移。上述各点均与癌的特点有所不同(表7-3)。正确掌握癌与肉瘤的特点,对临床诊断和治疗均有实际意义。
表7-3 癌与肉瘤的区别
| 癌 | 肉瘤 | |
| 组织来源 | 上皮组织 | 间叶组织 |
| 发病率 | 较常见,约为肉瘤的9倍,多见于40岁以上成人 | 较少见,大多见于青少年 |
| 大体特点 | 质较硬、色灰白、较干燥 | 质软、色灰红、湿润、鱼肉状 |
| 组织学特点 | 多形成癌巢,实质与间质分界清楚,纤维组织每有增生 | 肉瘤细胞多弥漫分布,实质与间质分界不清,间质内血管丰富,纤维组织少 |
| 网状纤维 | 癌细胞间多无网状纤维 | 肉瘤细胞间多有网状纤维 |
| 转移 | 多经淋巴道转移 | 多经血道转移 |
【常见的肉瘤有以下几种:】
1.纤维肉瘤(fibrosarcoma) 是肉瘤中常见的一种,其发生部位与纤维瘤相似,以四肢皮下组织为多见。分化好的纤维肉瘤细胞多呈梭形,异型性小,与纤维瘤有些相似;分化差的纤维肉瘤则有明显的异型性(图7-17)。纤维肉瘤分化好者生长慢,转移及复发较少见;分化差者生长快,易发生转移,切除后易复发。
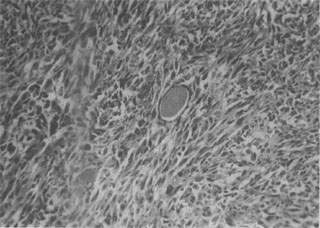
图7-17 纤维肉瘤
瘤细胞呈梭形,体积较大,核大,核分裂像较多,具明显的异型性
2.恶性纤维组织细胞瘤(malignant fibrous histiocytoma)这是近年来才受到重视的一种恶性肿瘤,为老年人最常见的软组织肉瘤。本瘤好发于下肢,其次是上肢的深部组织和腹膜后等处,也可发生于内脏器官,但较为少见。肿瘤细胞可有多种类型,电镜下,肿瘤细胞类型的多样性理更为明显,主要为纤维母细胞和组织细胞,此外尚见原始间叶细胞、肌纤维母细胞、黄色瘤细胞和多核瘤巨细胞。异型性往往十分明显,核分裂像多见。绝大多数肿瘤可见中等量到多量的慢性炎性细胞浸润。有的区域见纤维母细胞可呈束状交织排列和(或)排列成辐状,后者被认为有一定诊断价值;有的区域多形性明显,见多肿瘤细胞混杂分布,无一定排列形式而且异型性十分突出,可见形态坚异具有丰富嗜伊红胞浆的瘤巨细胞;有的区域粘液变性明显。关于此瘤的组织发生,有研究认为是由原始间叶细胞向不同方向分化所成。此瘤的恶性程度较高,切除后易复发和转移。
3.脂肪内瘤(liposarcoma) 为肉瘤中较常见的一种类型,多发生于大腿及腹膜后的软组织深部。来自原始间叶组织,极少从皮下脂肪层发生,与脂肪瘤的分布相反。这说明脂肪肉瘤极少是由脂肪瘤恶变而来,而是一开始即具恶性特征。本瘤多见于40岁以上成人,极少见于青少年。肉眼观,大多数肿瘤呈结节状或分叶状,表面常有一层假包膜,可似一般的脂肪瘤,亦可呈粘液性外观,或均匀一致呈鱼肉样。本瘤的瘤细胞形态多种多样,可见分化差的星形、梭形、小圆形或呈明显异型性和多形性的脂肪母细胞,胞浆内可见多少和大小不等的脂滴空泡(图7-18),也可见分化成熟的脂肪细胞,并常以某种细胞成分为主。间质有明显粘液性和大量血管网形成者,称为粘液样型脂肪肉瘤。当以分化差的小圆形脂肪母细胞为主(圆形细胞型脂肪肉瘤)或以多形性脂肪母细胞为主时(多形性脂肪肉瘤),恶性程度高,易有复发和转移。
图7-18 脂肪肉瘤
瘤细胞分散,胞浆内含大小不等的脂肪空泡。瘤细胞间有多量粘液性基质(图中呈灰色),并见瘤巨细胞(插入图)
4.横纹肌肉瘤(rhabdomyosarcoma)是较常见而且恶性程度很高的肉瘤。肿瘤由不同分化阶段的横纹肌母细胞组成。分化较高者红染的胞浆内可见纵纹和横纹,用磷钨酸苏木素染色更显而易见。根据瘤细胞的分化程度、排列结构和大体特点可分为三种类型:
(1)胚胎性横纹肌肉瘤:是最常见的一类,主要发生于10岁以下的婴幼儿和儿童。最好发的部位为头(眼眶、鼻腔、鼻咽部、中耳等处)、颈、泌尿生殖道及腹膜后。镜下见肿瘤由未分化和低分化的小圆或卵圆细胞、梭形或带状的横纹肌母细胞组成。以葡萄状息肉样肿物突出于粘膜为特点的葡萄状肉瘤(sarcoma botryoides)亦属此类型;
(2)腺泡状横纹肌肉瘤:常见于10~25岁的青少年,发生于四肢者较为多见。镜下特点为低分化的圆形或卵圆形瘤细胞形成不规则腺泡腔,在腺泡腔可偶见分化较高的横纹肌母细胞和多核瘤巨细胞。
(3)多形性横纹肌肉瘤:多见于成年人,好发于四肢的大肌肉,特别在大腿更为多见。镜下见瘤细胞呈明显异型性,可出现多种形态怪异的横纹肌母细胞,胞浆丰富红染,可见纵纹和横纹,核分裂像甚多(图7-2)。
各型横纹肌肉瘤均生长迅速,易早期发生血道转移,如不及时诊断治疗,预后极差,患者约90%以上在五年内死亡。
5.平滑肌肉瘤(leimyosarcoma)较多见于子宫及胃肠,偶可见于腹膜后、肠系膜、大网膜及皮下软组织。患者多为中老年人。平滑肌肉瘤的瘤细胞呈轻重不等的异型性,核分裂像的多少对判断其恶性程度有重要意义。恶性程度高者,手术后易复发,可经血道转移至肺、肝及其他器官。
6.血管肉瘤(hemangiosarcoma) 血管肉瘤起源于血管内皮细胞,可发生于各器官和软组织,发生于软组织者多见于皮肤,尤以头面部为多见。肿瘤多隆起于皮表,呈丘疹或结节状,暗红或灰白色。肿瘤内部易有坏死出血。有扩张的血管时,切面可呈海绵状。镜下,在分化较好者,瘤组织内血管腔形成明显,大小不一,形状不规则,被覆血管腔的内皮细胞大多有不同程度的异型性,可见核分裂像。分化差的血管肉瘤,细胞常呈片团状增生,形成不明显、不典型血管腔或仅呈裂隙状。瘤细胞异型性明显,核分裂像多见。血管肉瘤的恶性程度一般较高,常在淋巴结及肝、肺、骨等处形成转移。
7.骨肉瘤(osteosarcoma)和软骨肉瘤(chondrosarcoma)骨肉瘤起源于骨母细胞,软骨肉瘤起源于软骨细胞, 均为常见的骨恶性肿瘤(详见第十七章)。
第十节 神经外胚叶源性肿瘤
由神经外胚叶起源的肿瘤种类很多,有中枢神经系统和周围神经系统肿瘤(详见第十六章)、能分泌多肽激素及其前身的APUD系统来源的肿瘤(包括类癌、化学感受器瘤、嗜铬细胞瘤等,详见第十五章)、神网膜母细胞瘤、黑痣和黑色素瘤等。现仅将后三者分述如下。
1.视网膜母细胞瘤视网膜母细胞瘤(retinoblastoma)是来源于视网膜胚基的恶性肿瘤,绝大多数发生在3岁以内的婴幼儿,6岁以上罕见。7%在出生时即已存在。此瘤是一种常染色体显性遗传性疾病,并有家族史。大多数发生在一侧眼,但亦可在双眼发生。内眼观肿瘤为灰白色或黄色的结节状肿物,切面有明显的出血及坏死,并可见钙化点。肿瘤最初在视网膜上生长,以后向周围浸润:向前可侵入玻璃体,进而破坏眼球侵入眶内;向后侵入神经乳头,并可侵及视神经向眼球后和颅内蔓延。镜下见肿瘤由小圆形细胞构成,常只见核而胞浆不明显。核圆形、深染、核分裂像多见。有的瘤细胞停围绕一空腔作放射状排列,形成菊形团。转移一般不常见,发生时多循血路转移至骨、肝、肺、肾等处。淋巴道转移只在眼眶软组织被累及时才发生,多转移到耳前及颈淋巴结。预后一般不好,多在发病后一年半左右死亡。偶见自发性消退。
2.黑痣与黑色素瘤
(1)皮肤黑痣(pigmented nevus):来源于表基底层的黑色素细胞,为良性增生性病变,但有的可恶变成为黑色素瘤。根据其在皮肤组织内发生部位的不同,可分为交界痣(即痣细胞在表皮和真皮的交界处生长,形成多个细胞巢团,此型痣较易恶变为黑色素瘤),皮内痣(是最常见的一种,痣细胞在真皮内呈巢状或条索状排列)和混合痣(即同时有交界痣和皮内痣的改变)三种。
(2)黑色素瘤(melanmoa):又称为恶性黑色素瘤,是一种能产生黑色素的高度恶性肿瘤,大多见于30岁以上成人,发生于皮肤者以足底部和外阴及肛门周围多见,可以一开始即为恶性,但通常由交界痣恶变而来。凡黑痣色素加深、体积增大、生长加快或溃破、发炎和出血等常是恶变的象征。此瘤也可发生于粘膜和内脏器官。黑色素瘤的组织结构呈多样性,瘤细胞可呈巢状、条索状或腺泡样排列。瘤细胞可呈多边形或梭形,核大,常有粗大的嗜酸性核仁,胞浆内可有黑色素颗粒。也有胞浆内没有黑色素颗粒的黑色素瘤,称为无黑色素性黑色素瘤,但多巴反应可为阳性。电镜下,则可见其胞浆内含有少数典型的黑色素小体(melanosome)或前黑色素小体(premelanosome),有助于诊断。黑色素瘤的预后大多很差,晚期可有淋巴道及血道转移。因此,本瘤早期诊断和及时治疗十分重要。
第十一节 多种组织构成的肿瘤
有的肿瘤实质由两种以上不同类型组织构成,称为混合瘤。最复杂的混合瘤是畸胎瘤,基肿瘤实质由来源于三个胚层的各种组织混杂在一起构成,有如一个畸形的胎儿。还有肾胚胎瘤也是比较多样的一种肿瘤。癌肉瘤也属混合瘤之一种。现分述如下:
1. 畸胎瘤(teratoma)
畸胎瘤是来源于有多向分化潜能的生殖细胞的肿瘤,往往含有三个胚层的多种多样组织成分,排列结构错乱。根据其外观又可分为囊性及实性两种:根据其组织分化成熟程度不同,又可分为良性畸胎瘤和恶性畸胎瘤二类。本瘤最常发生于卵巢和睾丸。偶可见于纵隔、骶尾部、腹膜、松果体等部位。
(1)良性畸胎瘤:多为囊性,故也称为囊性畸胎瘤,或称为皮样囊肿,多见于卵巢。肿瘤多为单房性,内壁为颗粒体,粗糙不平,常有结节状隆起。有时能见到小块骨、软骨等,囊腔内有皮脂、毛发,甚至可见牙。镜下,除见皮肤组织及皮肤附件外,还可见到覆以立方上皮的腺体、气管或肠粘膜、骨、软骨、脑、平滑肌、甲状腺等组织。各种组织基本上分化成熟,故有成熟畸胎瘤之称。良性畸胎瘤预后好,少数可恶变为鳞状细胞癌。
(2)恶性畸胎瘤:多为实体性,在睾丸比卵巢多见。主要由分化不成熟的胚胎样组织组成,常见有分化不良的神经外胚层成分,故又称不成熟型畸胎瘤。本瘤常发生转移,可转移盆腔及远处器官。
2.肾胚胎瘤(embryonic tumor of kidney) 亦称肾母细胞瘤(nephroblastoma)、Wilms瘤。由肾内残留的未成熟胚胎组织发展而来,多见于5岁以下儿童。肿瘤成分多样,除见瘤细胞呈巢团状排列,形成幼稚的肾小球或肾小管样结构外,间质中可见疏松的粘液样组织,有时也可见到横纹肌、软骨、骨或脂肪组织(详见十二章)。
3.癌肉瘤(carcinosarcoma)同一肿瘤中既有癌又有肉瘤成分者称为癌肉瘤。癌的成分可为鳞状细胞癌、移行细胞癌、腺癌、分化差的癌等;肉瘤成分可分为纤维肉瘤、平滑肌肉瘤、横纹肌肉瘤、骨肉瘤、软骨肉瘤等。癌和肉瘤的成分可按不同比例混合,通常含癌和肉瘤成分各一种,偶尔不止一种,如腺癌与平滑肌肉瘤和骨肉瘤混合。癌肉瘤可以多种形式发生,如上皮组织和间叶组织同时发生恶变,多能干细胞向癌和肉瘤两种方向分化;癌诱导其间质发生恶变;癌细胞部分发生间叶组织化生和恶变等。
第十二节 肿瘤的病理学检查方法
肿瘤的病理学检查为极其重要的肿瘤诊断方法之一。病理学检查可以确定肿瘤的诊断、组织来源以及性质和范围等,为临床治疗提供重要的依据。肿瘤的病理学检查方法包括:
一、常规的病理形态学检查
1.我国医务工作者研制成食管细胞采取器(食管拉网法)检查食管癌及贲门癌(阳性确诊率为87.3%~94.2%)。还用鼻咽乳胶球细胞涂片、负压吸引细胞法及泡沫塑料海绵涂片法等采取鼻咽分泌物检查鼻咽癌,提高了阳性诊断率(阳性率为88%~92%)。用胃加压冲洗法采取胃内容物检查胃癌,也使阳性诊断率有了显着的提高。
2.活体组织检查从患者身体的病变部位取出小块组织(根据不同情况可采用钳取、切除或穿刺吸取等方法)或手术切除标本制成病理切片,观察细胞和组织的形态结构变化,以确定病变性质,作出病理诊断,称为活体组织检查(biopsy),简称活体。这是诊断肿瘤常用的而且较为准确的方法。近年来由于各种内窥镜(如纤维胃镜、纤维结肠镜、纤维支气管镜等)和影像诊断技术的不断改进,不但可以直接观察某些内肿瘤的外观形态,还可在其指引下准确地取材,进一步提高了早期诊断的阳性率。
二、新开展的检查方法
1.免疫组织化学检查免疫组化是最近10多年来迅速发展起来的一门新兴技术。它已被广泛运用肿瘤研究和诊断,其原理是利用抗原与抗体的特异性结合反应来检测组织中的未知抗原或者抗体,主要是肿瘤相关抗原(肿瘤分化抗原和肿瘤胚胎抗原),借以判断肿瘤的来源和分化程度,协助肿瘤的病理诊断和鉴别诊断。目前常用的染色方法有过氧化物酶-抗过氧化物酶法,即PAP法(peroxidaseantiperoxidase technique)和卵白素-生物素-过氧化物酶复合物法,即ABC法(avidin-biotin-peroxidase complex technique)。利用免疫组织化学方法已经可以对许多常规方法难以判断其来源的肿瘤加以鉴别。例如检测细胞骨架的中间丝(intermediate filament),其直径平均为10nm,介于微管和微丝之间。中间丝有五类:即神经原纤维、胶质原纤维酸性蛋白、结蛋白(desmin)、波形蛋白(vimentin)和角蛋白(keratin)。它们各有生物化学和免疫学特性,并分别存在于不同类型的细胞中,故具有相对的特异性,可用来协助诊断相应的神经细胞、神经胶质细胞、横纹肌和平滑肌、间叶组织和上皮细胞来源的肿瘤。利用激素和激素受体的特异性结合,还可以对乳腺癌等激素依赖性肿瘤的雌激素受体、孕激素受体的水平进行免疫组化测定。雌激素受体阳性者对于内分泌治疗的效果较好,预后也优于受体阴性的病人。
目前能用于肿瘤辅助诊断和鉴别诊断的抗体已不胜枚举。由于经验的积累,过去认为在诊断某些肿瘤上具有特异性的抗体也不是那样特异了。因此在判断结果时必须紧密地结合形态学和临床改变。
2.电子显微镜检查迄今尚未发现可据以诊断肿瘤和恶性肿瘤的特异性的超微结构改变。因此要鉴别是否为肿瘤和肿瘤的良恶性仍主要靠光镜观察。但电镜在确定肿瘤细胞的分化程度,鉴别肿瘤的类型和组织发生上可起重要作用。例如鉴别分化差的癌及肉瘤;区分各种恶性小圆细胞肿瘤,如神经母细胞瘤、Ewing肉瘤、胚胎性横纹肌肉瘤、恶性淋巴瘤及未分化小细胞癌。
在电镜下,癌细胞之间常见较多的桥粒连接或桥粒样连接,因而可与肉瘤相区别。在恶性小圆细胞肿瘤中,各类肿瘤也有其超微结构特点,如神经母细胞瘤常见大量树状细胞突,在瘤细胞体及胞突中均可查见微管和神经分泌颗粒;Ewing肉瘤的瘤细胞常分化差,胞浆内细胞器很少,但以大量糖原沉积为其特点:胚胎性横纹肌肉瘤可见由肌原纤维和Z带构成的发育不良的肌节;小细胞癌常可见细胞间连接和胞浆内神经分泌颗粒;恶性淋巴瘤除可见发育不同阶段淋巴细胞的超微结构特点外,不见细胞连接、神经分泌颗粒、树状胞突和糖原沉积,从而可与其他小圆细胞肿瘤区别。
3.流式细胞术(flow cytometry)流式细胞术是近年来发展起来的一种快速定量分析细胞的新技术,目前已广泛用于肿瘤研究,特别是应用于瘤细胞DNA含量的检测。许多资料表明,实体恶性肿瘤的DNA倍体大多为非整倍体或多倍体,所有良性病变都是二倍体。检测异常DNA含量不但可作为恶性肿瘤的标志之一,且可反映肿瘤的恶性程度及生物学行为。
4.图像分析技术病理形态学的观察基本上是定性的,缺乏精确而更为客观的定量标准。图像分析技术(image analysis)的出现弥补了这个缺点。随着电子计算机技术的发展,形态定量技术已从二维空间向三维空间发展。在肿瘤病理方面图像分析主要应用于核形态参数的测定(区别癌前病变和癌;区别肿瘤的良恶性;肿瘤的组织病理分级及判断预后等),DNA倍体的测定,显色反应(如免疫组织化学)的定量等方面。
5.分子生物学技术十余年来分子生物学肿瘤研究领域引起了一场革命。重组DNA技术、核酸分子杂交技术、聚合酶链反应(polymerase chain reaction,PCR)和DNA测序等新技术在肿瘤的基因分析和基因诊断上已经开始应用。例如对恶性淋巴瘤,利用Southern印迹杂交技术和PCR方法,可以对样本淋巴组织中是否存在单克隆性的增生做出判断,从而协助形态学诊断。这些技术还被用于肿瘤的病因和发病学研究。
第十三节 肿瘤的病因学和发病学
肿瘤病因学研究引起肿瘤的始动因素,肿瘤发病学则研究肿瘤的发病机制与肿瘤发生的条件。要治愈肿瘤和预防肿瘤的发生,关键问题是查明肿瘤的病因及其发病机制。
关于肿瘤的病因学和发病学,多年来进行了广泛的研究,虽然至今尚未完全阐明,但近年来分子生物学的迅速发展,特别是对癌基因和肿瘤抑制基因的研究,已经初步揭示了某些肿瘤的病因与发病机制,例如伯基特淋巴瘤和人类T细胞白血病/淋巴瘤。目前的研究表明,肿瘤从本质上说是基因病。引起遗传物质DNA损害(突变)的各种环境的与遗传的致癌因子可能以协同的或者序贯的方式,激活癌基因或(和)灭活肿瘤的抑制基因,使细胞发生转化(transformation)。被转化的细胞可先呈多克隆性增生,经过一个漫长的多阶段的演进过程(progression),其中一个克隆可相对无限制地扩增,通过附加突变,选择性地形成具有不同特点的亚克隆(异质性),从而获得浸润和转移的能力(恶性转化),形成恶性肿瘤。图7-19示肿瘤的病因和发病机制模式。
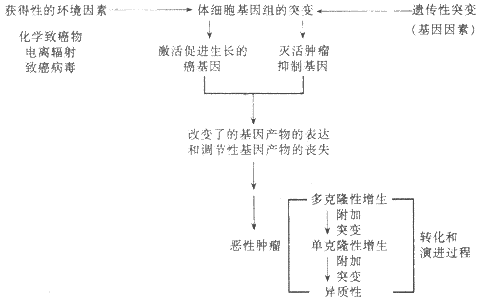
图7-19肿瘤的病因和发病机制模式图(采自Kumar,稍改)
一、肿瘤发生的分子生物学基础
1.癌基因
(1)原癌基因、癌基因及其产物:现代分子生物学的重大成就之一是发现了原癌基因(proto-oncogene)和原癌基因具有转化成致癌的癌基因(oncogene)的能力。Bishop和Varmus因为在这方面的贡献而获得1989年的诺贝尔奖。
癌基因是首先在逆转录病毒(RNA病毒)中发现的。含有病毒癌基因的逆转录病毒能在动物迅速诱发肿瘤并能在体外转化细胞。后来在正常细胞的DNA中也发现了与病毒癌基因几乎完全相同的DNA序列。被称为细胞癌基因。如ras,myc等。由于细胞癌基因在正常细胞中乃以非激活的形式存在,故又称为原癌基因。原癌基因可以由于多种因素的作用使其结构发生改变,而被激活成为癌基因。
原癌基因编码的蛋白质大多是对正常细胞生长十分重要的细胞生长因子和生长因子受体,如血小板衍生生长因子(PDGF)、纤维母细胞生长因子(FGF)、表皮细胞生长因子受体(EGF-R)、重要的信号转导蛋白质(如酪氨酸激酶、丝氨酶-苏氨酸激酶等)以及核调节蛋白(如转录激活蛋白)等。表7-4示常见的癌基因及其产物。
表7-4 几种常见的癌基因及其激活方式和相关的人类肿瘤
| 编码的蛋白质 | 原癌基因 | 激活机制 | 相关人类肿瘤 |
| 生长因子: | |||
| PDGF-β链 | sis | 过度表达 | 星形细胞瘤,骨肉瘤 |
| FGF | hst-1,int-2 | 过度表达 | 胃癌,膀胱癌,乳腺癌 |
| 生长因子受体: | |||
| EGF受体 | erb-B1 | 扩增 | 胶质瘤 |
| EGF样受体 | neu(erb-B2) | 扩增 | 乳腺癌,卵巢癌,肾癌 |
| 信号转导蛋白: | |||
| GTP-结合蛋白 | ras | 点突变 | 多种人体肿瘤,包括肺,结肠,胰,白血病 |
| 酪氨酸激酶 | abl | 易位 | 慢性粒细胞性,急性淋巴细胞性白血病 |
| 核调节蛋白: | |||
| 转录激活蛋白 | myc | 易位 | 伯基特淋巴瘤 |
| N-myc | 扩增 | 神经母细胞瘤,小细胞肺癌 | |
| 线粒体蛋白: | bcl-2 | 易位 | 滤泡性B-细胞淋巴瘤 |
(采自Basic Pathology;1992)
(2)原癌基因的激活:原癌基因在各种环境的或遗传的因素作用下,可发生结构改变(突变)而变为癌基因;也可以是原癌基因本身结构没有改变,而是由于调节原癌基因表达的基因发生改变使原癌基因过度表达。以上基因水平的改变可继而导致细胞生长刺激信号的过度或持续出现,使细胞发生转化。引起原癌基因突变的DNA结构改变包括点突变(如90%的胰腺癌有ras 基因的点突变)、染色体易位(如伯基特淋巴瘤的t(8:14),慢性粒细胞白血病的Phl染色体)、插入诱变、基因缺失和基因扩增(如神经母细胞瘤的N-myc原癌基因可复制成多达几百个拷贝,在细胞遗传学上表现为染色体出现双微小体和均染区)。癌基因编码的蛋白质(癌蛋白)与原癌基因的正常产物相似,但有质或量的不同。通过生长因子或生长因子受体增加、产生突变的信号转导蛋白与DNA结合的转录因子等机制,癌蛋白调节其靶细胞的代谢、促使该细胞逐步转化,成为肿瘤细胞。
2.肿瘤抑制基因与原癌基因编码的蛋白质促进细胞生长相反,在正常情况下存在于细胞内的另一类基因——肿瘤抑制基因的产物能抑制细胞的生长。若其功能丧失则可能促进细胞的肿瘤性转化。由此看来,肿瘤的发生可能是癌基因的激活与肿瘤抑制基因的失活共同作用的结果。目前了解最多的两种肿瘤抑制基因是Rb基因和P53基因。它们的产物都是以转录调节因子的方式控制细胞生长的核蛋白。其它肿瘤抑制基因还有神经纤维瘤病-1基因、结肠腺瘤性息肉基因、结肠癌丢失基因和Wilms瘤-1等。
(1)Rb基因:Rb基因随着对一种少见的儿童肿瘤——视网膜母细胞瘤的研究而最早发现的一种肿瘤抑制基因。Rb基因的纯合子性的丢失见于所有的视网膜母细胞瘤及部分骨肉瘤、乳腺癌和小细胞肺癌等。Rb基因定位于染色体13q14,编码一种核结合蛋白质(P105-Rb)。它在细胞核中以活化的脱磷酸化和失活的磷酸化的形式存在。活化的Rb蛋白对于细胞从G0/G1期进入S期有抑制作用。当细胞受到刺激开始分裂时,Rb 蛋白被磷酸化失活,使细胞进入S期。当细胞分裂成两个子细胞时,失活的(磷酸化的)Rb蛋白通过脱磷酸化再生使子细胞处于G1期或G0的静止状态。如果由于点突变或13q14的丢失而使Rb基因失活,则Rb蛋白的表达就会出现异常,细胞就可能持续地处于增殖期,并可能由此恶变。
(2)p53基因:p53基因定位于17号染色体。正常的p53蛋白(野生型)存在于核内,在脱磷酸化时活化,有阻碍细胞进入细胞周期的作用。在部分结肠癌、肺癌、乳腺癌和胰腺癌等均发现有p53基因的点突变或丢失,从而引起异常的p53蛋白表达,而丧失其生长抑制功能,从而导致细胞增生和恶变。近来还发现某些DNA病毒,例如HPV和SV-40,其致癌作用是通过它们的癌蛋白与活化的Rb蛋白或p53蛋白结合并中和其生长抑制功能而实现的。
3.多步癌变的分子基础恶性肿瘤的发生是一个长期的、多因素造成的分阶段的过程,这已由流行病学、遗传学和化学致癌的动物模式所证明。近年来的分子遗传学研究从癌基因和肿瘤抑制基因的角度为此提供了更加有力的证明。单个基因的改变不能造成细胞的完全恶性转化,而是需要多基因的改变,包括几个癌基因的激活和两个或更多肿瘤抑制基因的丧失。以结肠癌的发生为例,在从结肠上皮过度增生到结肠癌的演进过程中,关键性的步骤是癌基因以及肿瘤抑制基因的丧失或突变。这些阶段梯性积累起来的不同基因分子水平的改变,可以在形态学的改变上反映出来(图7-20)。
图7-20 结直肠癌通过上皮增生-腺瘤-癌的阶梯性的演进的分子生物学和形态学改变的关系(采自Kumar)
二、环境致癌因素及其致癌机制
1.化学致癌因素现已确知的对动物有致癌作用的化学致癌物约有1000多种,其中有些可能和人类癌瘤有关。对化致癌物的研究表明:①各种化学致癌物在结构上是多种多样的。基中少数不需在体内进行代谢转化即可致癌,称为直接作用的化学致癌物,如烷化剂。绝大多数则只有在体内(主要是在肝)进行代谢,活化后才能致癌,称为间接作用的化学致癌物或前致癌物,其代谢活化产物称终末致癌物。如3,4-苯并芘是间接致癌物,其终末致癌物是环氧化物;②所有的化学致癌物在化学上都具有亲电子结构的基团,如环氧化物、硫酸酯基团等。它们都与细胞大分子的亲核基团(如DNA分子中的鸟嘌呤的N-7、C-8,腺嘌呤的N-1、N-3,胞嘧啶的N-3等)共价结合,形成加合物,导致DNA的突变。化学致癌物大多数是致突变剂(mutagens);③某些化学致癌物的致癌作用可由其它无致癌作用的物质协同作用而增大。这种增加致癌效应的物质称为促癌物(promoter),如巴豆油、激素、酚和某些药物。致癌物引发初始变化称为激发作用(initiation),而促癌物的协同作用称为促进作用(promotion)。据此,Berenblum(1942)提出致癌过程的第二阶段学说,即激发和促进两个过程。现在认为激发过程是由致癌物引起的不可逆的过程,使得一种原癌基因(如ras基因)突变性活化,这种突变可遗传给子代细胞;促进过程据目前研究,可能是由于促癌剂(如巴豆油)是细胞内信号转导通道的关键性成分——蛋白激酶C的活化剂,并且能使某些细胞分泌生长因子所致。因此促进作用能促使突变的细胞克隆性生长、抑制其正常分化,最后在附加突变的影响下形成恶性肿瘤。此学说在预恶性肿瘤具有现实意义,因为激发过程是很短暂的,大多不可逆转,而促进过程则很长,一般需10~20年。因此,如能减少环境中的促癌因子,亦可有效地预防恶性肿瘤的发生。
主要的化学致癌物质有以下几类:
(1)间接作用的化学致癌物:
1)多环芳烃:存在于石油、煤焦油中。致癌性特别强的有3,4-苯并芘,1,2,5,6-双苯并蒽,3-甲基胆蒽及9,10-二甲苯蒽等。这些致癌物质在使用小剂量时即能在实验动物引起恶性肿瘤,如涂抹皮肤可引起皮肤癌,皮下注射可引起纤维肉瘤等。3,4-苯并芘是煤焦油的主要致癌成分,还可由于有机物的燃烧而产生。它存在于工厂排出的煤烟、烟草点燃后的烟雾中。近几十年来肺癌的发生率之日益增加,公认与吸烟和工业城市严重的大气污染有密切关系。此外,据调查,烟熏和烧烤的鱼、肉等食品中也含有多环芳烃,这可能和某些地区胃癌的发病率较高有一定关系。多环芳烃在肝经细胞色素氧化酶P450系统氧化成环氧化物,后者以其亲电子基因(不饱和的C-C键)与核酸分子以共价健结合而引起突变。
2) 芳香胺类与氨基偶氮染料:致癌的芳香胺类,如乙荼胺、联苯胺、4-氨基联苯等,与印染厂工人和橡胶工人的膀胱癌发生率较高有关。氨基偶氮染料,如以前在食品工业中曾使用过的奶油黄(二甲基氨基偶氮苯,可将人工奶油染成黄色的染料)和猩红,在动物实验可引起大白鼠的肝细胞性肝癌。以上两类化学致癌物主要在肝代谢。芳香胺的活化是在肝通过细胞色素氧化酶P450系统使其N端羟化形成羟胺衍生物,然后与葡萄糖醛酸结合成葡萄糖苷酸从泌尿道排出,并在膀胱水解释放出活化的羟胺而致膀胱癌。
3)亚硝胺类:亚硝胺类物质致癌谱很广,可在许多实验动物诱发各种不同器官的肿瘤。但是近年来引起很大兴趣的主要是可能引起人体胃肠癌或其它肿瘤。亚硝酸盐可作为肉、鱼类食品的保存剂与着色剂进入人体;也可由细菌分解硝酸盐产生。在胃内的酸性环境下,亚硝酸盐与来自食物的各种二级胺合成亚硝胺。我国河南林县的流行病学调查表明,该地食管癌发病率很高与食物中的亚硝胺高含量有关。亚硝胺在体内经过羟化作用而活化,形成有很强的反应性的烷化碳离子而致癌。
4)真菌毒素:黄曲霉菌广泛存在于高温潮湿地区的霉变食品中,尤以霉变的花生、玉米及谷类含量最多。黄曲霉毒素有许多种,其中黄曲霉毒素B1(aflatoxin B1)的致癌性最强,据估计其致癌强度比奶油黄大900倍,比二甲基亚硝胺大75倍,而且化学性很稳定,不易被加热分解,煮熟后食入仍有活性。黄曲霉毒素B1的化学结构为异环芳烃,在肝通过肝细胞内的混合功能氧化酶氧化成环氧化物而致突变。这种毒素主要诱发肝细胞性肝癌。我国和南非肝癌高发区的调查都显示黄曲霉毒素B1在食物中的污染水平与肝癌的发病率有关。但这些地区同时也是乙型肝炎病毒(HBV)感染的高发区。在HBV感染与黄曲霉毒素B1污染之间的关系方面,分子生物学的研究表明,黄曲霉毒素B1的致突变作用是使肿瘤抑制基因p53发生点突变而失去活性,而HBV感染所致的肝细胞慢性损伤和由此引起的肝细胞持续再生为黄曲霉毒素B1的致突变作用提供了有利的条件。因此HBV感染与黄曲霉毒素B1的协同作用是我国肝癌高发地区的主要致癌因素。此外,也已证明,在我国食管癌高发地区居民食用的酸菜中分离出的白地霉菌,其培养物有促癌或致癌作用。
(2)直接作用的化学致癌物:这类化学致癌物不需要体内代谢活化即可致癌,一般为弱致癌剂,致癌时间长。
1)烷化剂与酰化剂:例如抗癌药中的环磷酰胺、氮芥、本丁酸氮芥、亚硝基脲等。这类具有致癌性的药物可在应用相当长时间以后诱发第二种肿瘤,如在化学治疗痊愈或已控制的白血病、何杰金淋巴瘤和卵巢癌的病人,数年后可能发生第二种肿瘤,通常是粒细胞性白血病。某些使用烷化剂的非肿瘤病人,如类风湿性关节炎和Wegener肉芽肿的病人,他们发生恶性肿瘤的机率大大高于正常人。因此这类药物应谨慎使用。
2)其它直接致癌物:金属元素对人类也有致癌的作用,如镍、铬、镉、铍等,如炼镍工人中,鼻癌和肺癌明显高发;镉与前列腺癌、肾癌的发生有关;铬可引起肺癌等。其原因可能是金属的二价阳离子,如镍、镉、铅、铍、钴等,是亲电子的,因此可与细胞大分子,尤其是DNA反应。例如镍的二价离子可以使多聚核苷酸解聚。一些非金属元素和有机化合物也有致癌性,如砷可诱发皮肤癌;氯乙烯可致塑料工人的肝血管肉瘤;苯致白血病等,也受到关注。
化学致癌大多与环境污染和职业因素有关,因此彻底治理环境污染,防治职业病,对于减少恶性肿瘤的发病是极其重要的。目前发现的具有防癌或抗癌作用的稀有元素有钼、硒、镁、铂等。表7-5列出主要的化学致癌物、易感人群和诱发的肿瘤。
表7-5 主要的化学致癌物及易感人群和诱发的肿瘤
| 化学致癌物 | 易感人群 | 诱发的主要肿瘤 |
| 直接作用的 | ||
| 烷化剂 | 接受化学治疗的恶性肿瘤病人 | 白血病 |
| 间接作用的 | ||
| 多环芳烃 | 吸烟者、食用熏制鱼肉者 | 肺癌、胃癌 |
| 芳香胺 | 染料工人、橡胶工人 | 膀胱癌 |
| 亚硝胺 | 亚硝酸盐污染食物的食用者 | 食管癌、胃癌 |
| 黄曲霉毒素B1 | 污染食物的食用者 | 肿细胞性肿癌 |
| 石棉纤维 | 矿工、接触者 | 肺癌、胸膜间皮瘤 |
| 氯乙烯 | 塑料厂工人 | 肝血管肉瘤 |
| 苯 | 橡胶工人、染料工人 | 白血病 |
| 砷 | 矿工、农药工人和喷撒者 | 皮肤癌、肺癌、肝癌 |
| 镍 | 炼镍工人 | 鼻癌、肺癌 |
| 铬 | 接触含铬气体者 | 鼻癌、肺癌、喉癌 |
| 镉 | 接触者 | 前列腺癌、肾癌 |
(采自Basic Pathology,1992,有改动)
2.物理性致癌因素已证实的物理性致癌因素主要是离子辐射。异物、慢性炎性刺激和创伤亦可能与促癌有关。
(1)电离辐射:包括X射线、γ射线、亚原子微粒(β粒子、质子、中子或α粒子)的辐射以及紫外线照射。大量事实证明,长期接触X射线及镭、铀、氡、钴、锶等放射性同位素,可以引起各种不同的恶性肿瘤,例如放射工作者长期接触X射线而又无必要的防护措施时,常可发生手部放射性皮炎以致皮肤癌;其急性和慢性粒细胞性白血病的发生率亦较一般人高10倍以上。在出生前或出生后接受过X线照射的儿童,其急性白血病的发生率高于一般儿童。开采含放射性物质(钴、氡等)的矿工易患肺癌。日本长崎、广岛在第二次世界大战时受原子弹爆炸影响的幸存居民,经过长期观察,发现慢性粒细胞白血病的发生率明显增高(照射后4~8年为发病高峰),甲状腺癌、乳腺癌、肺癌等的发生率亦较高。在婴幼儿期接受过颈部放射线照射者,甲状腺癌发生率明显增高。有些放射性同位素如32P、48Sr、210Po、239Pu等摄入能诱发骨肉瘤。
辐射能使染色体断裂、易位和发生点突变,因而激活癌基因或者灭活肿瘤抑制基因。由于与辐射有关的肿瘤的潜伏期较长,因此肿瘤最终可能当辐射所损伤的细胞的后代又受到其它环境因素(如化学致癌剂、病毒等)所致的附加突变之后,才会出现。
动物实验和临床观察均证实,阳光中紫外线长期过度照射可引起外露皮肤的鳞状细胞癌、基底细胞癌和恶性黑色素瘤。白种人或照射后色素不增加的有色人种最易发生。其作用机制是细胞内DNA吸收了光子,使其中相邻的两个嘧啶连接(包括胸腺嘧啶与胸腺嘧啶、胸腺嘧啶与胞嘧啶、胞嘧啶与胞嘧啶),形成嘧啶二聚体。二聚体又形成环丁烷,从而破坏DNA双螺旋中二聚体所在处的磷酸二酯骨架,妨碍DNA分子的复制。在正常人这种损害通常可为一系列DNA修复酶所修复,因此皮肤癌发病少见。而一种罕见的常染色体隐性遗传病——着色性干皮病(xeroderma pigmentosum)的患者,由于先天性缺乏修复DNA所需的酶,不能将紫外线所致的DNA的损害修复,皮肤癌的发病率很高。
(2)热辐射的促癌作用:克什米尔人冬季习惯用怀炉取暖,有时在腹部引起“怀炉癌”;我国西北地区居民冬季烧火取暖,有时臀部皮肤发生癌变形成所谓“炕癌”。这些说明长期的热辐射可能有一定的促癌作用。在烧伤瘢痕的基础上易发生“瘢痕癌”,有人在烧伤瘢痕中发现化学致癌物。
(3)慢性炎性刺激:肿瘤必须在细胞增生的基础上发生。慢性炎症时产生的细胞生长因子能使细胞持续增生,在此基础上DNA易发生突变而发生肿瘤。因而慢性刺激有促癌作用。慢性皮肤溃疡、结石引起的慢性胆囊炎、慢性子宫颈炎和子宫内膜增生等病变有时可发生癌变,可能即与此有关。
(4)异物:石棉和石棉制品能导致人的胸膜间皮瘤,重度暴露于石棉纤维的工人,其胸膜间皮瘤的发生率可达2%~3%,潜伏期一般为20年。动物实验证明,植入动物体内的异物,如塑料、金属片,玻璃纤维等,可以诱发各种肉瘤。肿瘤的发生与植入物体的化学性关系不大而与物体表面的形状、光滑程度和耐久性有关。人类对于异物刺激有较大的耐受性,虽然尚无植入塑料和金属致癌的报道,但是已人工乳房(有机硅橡胶制成)与乳腺癌有关的报告。
另一种与肿瘤有关的异物是寄生虫。医学文献中早就有埃及血吸虫感染的病人,其膀胱癌的发生率高于正常人的报告。在我国,日本血吸虫
■[此处缺少一些内容]■
RNA病毒(逆转录病毒)的研究导致了癌基因的发现,并由此开创了肿瘤的分子遗传学。在人类已知的与肿瘤有关的病毒并不多。
(1)RNA致瘤病毒:对动物逆转录病毒致癌的研究发现,由于病毒类型的不同,它们是通过转导(transduction)或插入突变(insertional mutagenesis)这两种机制将其遗传物质整合到宿主细胞DNA中,并使宿主细胞分生转化的。①急性转化病毒:这类病毒含有从细胞的原癌基因转导的病毒癌基因,如src,abl,myb等,这些病毒感染细胞后,将以其病毒RNA为模板通过逆转录酶合成的DNA片断整合(integration)到宿主的DNA链中并表达,导致细胞的转化;②慢性转化病毒:这类病毒(如鼠乳腺癌病毒)本身并不含有癌基因,但是有促进基因,当感染宿主细胞后促进基因也可由于逆转录酶的作用而插入到宿主细胞DNA链中的原癌基因附近,引起正常的或突变的原癌基因激活并且过度表达,使宿主细胞转化。
人类T细胞白血病/淋巴瘤病毒I(human T-cell leukemia/lymhoma virus I,HTVL-1)是与人类肿瘤发生密切相关的一种RNA病毒,与主要流行于日本和加勒比地区的T细胞白血病/淋巴瘤有关。HTLV-1病毒与AIDS病毒一样,转化的靶细胞是CD4+的T细胞亚群(辅助T细胞)。HTLV-1在人类是通过性交、血液制品和哺乳传播的。受染人群发生白血病的机率为1%,潜伏期为20~30年。HTLV-1转化T细胞的机制还不甚清楚。HTLV-1不含有任何已知的癌基因,也未发现其在某一原癌基因附近的固定的整合位置。HTLV-1的转化活性与其RNA中的一个称为Tax的基因有关。Tax的产物对病毒的复制十分重要,因其通过对5’-长末端重复片段(5’-long terminal repeat region,5’-LTR)的作用刺激病毒mRNA的转录。Tax蛋白也可激活几种能引起T细胞增生的宿主基因的转录,如编码调节细胞内其它基因表达的P55蛋白c-fos基因,编码PDGF的c-sis基因,编码IL-2及其受体的基因和髓样生长因子(即粒-单核细胞集落刺激因子,GM-CSF)的基因。IL-2及其受体的基因激活后可以建立起一个自分泌体系(autocrine system)能直接引起T细胞的增生;GM-CSF作用于巨噬细胞,使其产生IL-1,从而引起T细胞的增生。因此HTLV-1是通过Tax基因转化细胞的。这些增生的T细胞最初是多克隆性的,而且出现二次突变的可能性大大增加,如其中的某一个发生第二次突变,将导致单克隆性的T细胞肿瘤。
(2)DNA致瘤病毒:DNA病毒中有50多种可引起动物肿瘤。对它们的研究,尤其是对多瘤病毒的研究,提示了DNA病毒致癌的机制。DNA病毒感染细胞后出现两种后果:①如果病毒DNA未能被整合到宿主的基因组中,病毒的复制不会受到干扰,大量的病毒复制最终使细胞死亡;②要引起细胞的转化,病毒基因必需整合到宿主的DNA中并且作为细胞的基因加以表达。多瘤病毒的T基因编码的蛋白质T抗原具有酪氨酸激酶活性,能像生长因子受体那样刺激细胞DNA合成,并使细胞持续增生,而后形成肿瘤。与人类肿瘤发生密切有关的DNA病毒有以下三种:
1)人类乳头状瘤病毒(human papilloma virus,HPV):HPV与人类上皮性肿瘤,主要是与子宫颈和肛门生殖器区域的鳞状细胞癌的关系,近年来已有大量资料予以证实。HPV的某些亚型(如16、18型)的DNA序列已在75%~100%的宫颈癌病例的癌细胞中发现。HPV的致癌机制还不完全清楚。近来发现HPV的16、18和31高危亚型的早期病毒基因产物E6和E7蛋白,极易与Rb和p53基因的产物结合并中和其抑制细胞生长的功能。在体外,Rb和p53基因产物的失活能使人类棘细胞转化并且长期存活,但不形成肿瘤。这时如果再转染一个突变的ras基因,就会引起完全的恶性转化。这说明HPV在致癌时不是单独作用的,需要环境因素的协同。
2)Epstein-Barr病毒(EBV):与之有关的人类肿瘤是伯基特淋巴瘤和鼻咽癌。
伯基特淋巴瘤是一种B细胞性的肿瘤。流行于非洲东部和散发于世界各地。在流行地区,所有病人的瘤细胞都携带EBV的基因组成分并且出现特异的染色体易位t(8:14)。EBV对B细胞有很强的亲和性,能使受染的B细胞发生多克隆性的增生。在正常的个体这种增生是可以控制的,受染者没有症状或者临床表现为自限性的传染性单核细胞增生症。而在非洲流行区,由于疟疾或其它感染损害了患者的免疫功能,受染B细胞乃持续增生。在此基础上如再发生附加的突变(如t(8:14)),则后者使c-myc激活,导致进一步的生长控制丧失,并在其它附加基因损伤的影响下,最终导致单克隆性的肿瘤出现。
鼻咽癌在我国南方和东南亚流行,EBV的基因组也在几所有肿瘤细胞中发现。但是与伯基特淋巴瘤一样,EBV在鼻咽癌发生中的作用也需要其它因素的配合。
3)乙型肝炎病毒(hepatitis b virus,HBV):流行病学调查发现,慢性HBV感染与肝细胞性肝癌的发生有密切的关系。但是HBV本身并不含有可以编码任何转化蛋白(癌蛋白)的基因,其中肝细胞DNA中的整合也没有固定的模式。HBV的致癌作用看来是多因素的:①如在前文所述,HBV导致的慢性肝损伤使肝细胞不断再生,这使另外的致癌因素(如黄曲霉毒素B1)的致突变作用容易发生;②HBV可能编码一种称为X蛋白的调节成分,使受染肝细胞的几种原癌基因激活;③在某些病人,HBV的整合可导致p53基因的失活。由此可见,肝细胞性肝癌的发生也可能是多步骤的。
三、影响肿瘤发生、发展的内在因素及其作用机制
肿瘤发生和发展是一个十分复杂的问题,除了外界致癌因素的作用外,机体的内在因素也起着重要作用,后者包括宿主对肿瘤反应,以及肿瘤对宿主的影响。这些内在因素是复杂的,许多问题至今尚未明了,还有待进一步研究。机体的内在因素可分为以下几方面;
1.遗传因素人类肿瘤是否有遗传性,以及遗传因素在人类肿瘤的发生上究竟起多大的作用?这是人们普遍关注并不断深入研究的课题。根据一些高癌家族系谱的分析,有以下几种不同情况:
(1)呈常染色显性遗传的肿瘤:如视网膜母细胞瘤、肾母细胞瘤、肾上腺或神经节的神经母细胞瘤等。一些癌前疾病,如结肠多发性腺瘤性息肉症、神经纤维瘤病等本身不是恶性肿瘤,但恶变率极高,有100%的结肠家族性多发性腺瘤性息肉病的病例在50岁以前发生恶变,成为多发性结肠腺癌。这些肿瘤和癌前疾病都属单基因遗传,以常染色体显性遗传的规律出现。其特点为早年(儿童期)发病,肿瘤呈多发性,常累及双侧器官。
(2)呈常染色体隐性遗传的遗传综合征:如患Bloom综合征(先天性毛细血管扩张性红斑及生长发育障碍)时易发生白血病及其他恶性肿瘤;毛细血管扩张性共济失调症患者多发生急性白血病和淋巴瘤;着色性干皮病患者经紫外光照射后易患皮肤基底细胞癌、磷状细胞癌或黑色素瘤。这些肿瘤易感性高的人常伴有某种遗传缺陷,如免疫缺陷、染色体缺陷和内切酶等的缺陷。
(3)遗传因素与环境因素在肿瘤发生中起协同作用,而环境因素更为重要,决定这类肿瘤的遗传因素是属于多基因的。目前发现不少常见肿瘤有家族史,如乳腺癌、胃肠癌、食管癌、肝癌、鼻咽癌、白血病、子宫内膜癌、前列腺癌、黑色素瘤等。
总的说来不同的肿瘤可能有不同遗传传递方式,真正直接遗传的只是少数不常见的肿瘤。遗传因素在大多数肿瘤发生中的作用是对致癌因子的易感性或倾向性。Kundson(1974)提出二次突变假说(two hit hypothesis)来解释遗传性损害在肿瘤发生中作用。以现代分子生物学的术语来描述这一假说是:以视网膜母细胞瘤例,Rb基因定位于染色体13q14,只有两条同源染色体上的Rb等位基因都被灭活,即需经两次突变后,才能使肿瘤发生。在家族性视网膜母细胞瘤患儿基因组中已经存在一个从父母得到的有缺陷的Rb基因拷贝,另一个Rb基因拷贝正常的(杂合型),因而只要再有一次体细胞突变,即可形成肿瘤(纯合型)。这种家族性视网膜母细胞瘤的患儿年龄小,双侧发病的较多。而在散发性的视网膜母细胞瘤的患儿,由于其两个正常的Rb等位基因都要通过体细胞突变而失活才能发病,故出现这种病例的可能性只有家族性的万分之一,而且发病较晚,多为单侧。
2.宿主对肿瘤的反应——肿瘤免疫恶性转化是由于遗传基因的改变引起的。有些异常基因表达的蛋白可引起免疫系统的反应,从而使机体能消灭这些“非已”的转化细胞。如果没有这种免疫监视机制,则肿瘤的发生要比实际上出现的多得多。关于肿瘤免疫的研究不仅对肿瘤的发生有重要的意义。而且为肿瘤的免疫治疗指出了方向。
(1)肿瘤抗原:引起机体免疫反应的肿瘤抗原可分为两类:①只存在于肿瘤细胞而不存在于正常细胞的肿瘤特异性抗原;②存在于肿瘤细胞和某些正常细胞的肿瘤相关抗原。
尽管在肿瘤特异性抗原的研究上花费了大量的时间和精力,企图寻找某种肿瘤的特异性抗原。但是现已在化学致癌的动物模型中发现,肿瘤特异性抗原是个体独特的,即不同个体中同一种致癌物诱发的同一组织学类型的肿瘤有不同的特异性抗原。因此用检测某种肿瘤特异性抗原来诊断或用某抗体来治疗某些肿瘤的可能性在目前尚不存在。肿瘤特异性抗原的个体独特性的原因是,癌变时癌基因发生突变的随机性引起异常蛋白的随机出现,因而无法产生特定的针对某一类肿瘤的抗原。
肿瘤相关抗原在肿瘤中的表达,推测与遗传因素的改变有关。它们又可分为两类:肿瘤胚胎抗原和肿瘤分化抗原。前者在正常情况下出现在发育中的胚胎组织而不见于成熟组织,但可见于癌变组织。例如在胚胎肝细胞和肝细胞性肝癌中出现的甲胎蛋白以及在胚胎组织和结肠癌中出现的癌胚抗原。后者是指肿瘤细胞具有的与分化程度有关的某些抗原。例如前列腺特异抗原见于正常前列腺上皮和前列腺癌细胞。肿瘤相关抗原在有关肿瘤的诊断上是有用的标记,也可用此制备抗体,用于肿瘤的免疫治疗。
(2)抗肿瘤的免疫效应机制:肿瘤免疫反应以细胞免疫为主,体液免疫为辅。参加细胞免疫的效应细胞主要有细胞毒性T细胞(CTL)、自然杀伤细胞(NK)和巨噬细胞。抗肿瘤免疫的机制如图7-21所示。CTL被白细胞介素2(IL-2)激活后可以通过其T细胞受识别瘤细胞上的人类主要组织相容性复合体(major histocompatibility compolex,MHC)I型分子而释放某些溶解酶将瘤细胞杀灭。CTL的保护作用在对抗病毒所致的肿瘤(如EBV引起的伯基特淋巴瘤和HPV导致的肿瘤)时特别明显。NK细胞是不需要预先致敏的、能杀伤肿瘤细胞的淋巴细胞。由IL-2激活后,NK细胞可以溶解多种人体肿瘤细胞,其中有些并不引起T细胞的免疫反应,因此NK细胞是抗肿瘤免疫的第一线的抵抗力量。NK细胞识别靶细胞的机制可能是通过NK细胞受体和抗体介导的细胞毒作用(antibody-dependent cellular cytotoxicity,ADCC)。巨噬细胞在抗肿瘤反应中是与T细胞协同作用的:T细胞产生的α-干扰素可激活巨噬细胞,而巨噬细胞产生的肿瘤坏死因子(TNF-α)和活性氧化代谢产物在溶解瘤细胞中起主要作用。此外巨噬细胞的Fc受体还可与肿瘤细胞表面的IgG结合,通过ADCC杀伤肿瘤细胞。体液免疫参加抗肿瘤反应的机制主要是激活补体和介导NK细胞参加的ADCC。
图7-21 抗肿瘤免疫的细胞效应机制
CTL:细胞毒性T淋巴细胞 NK:自然杀伤细胞 IFN-γ:γ-干扰素 TNF-α:肿瘤坏死因子α IL-2:白细胞介素2 MHCI:主要组织相容性复合体Ⅰ型分子
(3)免疫监视:免疫监视机制在抗肿瘤中的作用的最有力的证据是,在免疫缺陷病患者和接受免疫抑制治疗的病人中,恶性肿瘤的发病率明显增加。先天性免疫缺陷病(如X-性联无γ球蛋白血症)的病人有5%发生恶性肿瘤,这比对照组高出200倍。在器官移植的受者和AIDS患者中发生淋巴瘤的可能也大大增加。恶性肿瘤患者随着病程的发展和病情恶化每伴有免疫功能普遍下降,这在晚期患者尤为突出。相反,有些肿瘤,如神经母细胞瘤、恶性黑色素瘤和绒毛膜上皮癌等肿瘤患者,由于机体免疫功能增高,肿瘤可发生自发消退。但大多数恶性肿瘤乃发生于免疫功能正常的人群,这些肿瘤能逃脱免疫系统的监视并破坏机体的免疫系统,其机制还不甚清楚,可能与下列因素有关:①在肿瘤生长过程中,具有较强抗原性的亚克隆被免疫系统消灭,而无抗原性的或者抗原性弱的亚克隆则生长成肿瘤;②CTL攻击肿瘤细胞时要识别瘤细胞膜上的Ⅰ型MHC抗原,故肿瘤细胞MHC抗原表达的丧失或减少,会使瘤细胞避开CTL的攻击;③许多致癌因素同时也抑制宿主的免疫反应,如化学致癌物和电离辐射等;④肿瘤产物也可以抑制免疫反应,如由许多肿瘤分泌的转化生长因子β(TGF-β)就是一种潜在免疫抑制剂;⑤肿瘤引发的有些免疫反应,如抑制性T细胞的激活,本身就可抑制对肿瘤的免疫反应。
(4)肿瘤的免疫治疗:免疫治疗旨在通过替换免疫系统中的受到抑制的成分,或者刺激内源性的反应来增加机体的抗肿瘤能力。目前有三种主要的方法。①继承性细胞治疗:先将病人的周围血淋巴细胞与IL-2一起孵育,然后将产生的淋巴激肽激活杀伤细胞(lymphokine-activated killer cell,LAK细胞,主要由NK细胞组成)再输回病人;或者将从病人切除的肿瘤组织中的淋巴细胞(主要是CTL)在体外与IL-2一起培养后再输回病人。这两种治疗方法的效果还有待于评价;②细胞激肽治疗:主要是用IL-1,IFN-α、γ、TNF-α等细胞激肽与其它治疗方法一起用于抗癌,目前疗效最好的是INF-α对毛细胞白血病的治疗;③抗体治疗:目前的兴趣集中在用抗肿瘤相关抗原的单克隆抗体与细胞毒素结合后制成的“免疫毒素”来治疗肿瘤,这种“魔弹”的疗效正在观察中。
3.其它与肿瘤发病学有关的因素
(1)内分泌因素:内分泌紊乱与某些器官肿瘤的发生发展有密切的关系,如乳腺癌的发生发展可能与患者体内雌激素水平过高或雌激素受体的异常有关。乳腺癌在妊娠期和哺乳期发展得特别快,切除卵巢或用雌激素治疗可使肿瘤明显缩小。此外,激素与恶性肿瘤的转移及扩散也有一定的关系,如垂体前叶激素可促进肿瘤的生长和转移;肾上腺皮质激素对某些造血系统的恶性肿瘤有抑制其生长和转移的作用。
(2)性别和年龄因素:肿瘤的发生在性别上有很大的差异,除生殖器官肿瘤及乳腺癌在女性明显较多见外,胆囊、甲状腺及膀胱等器官的肿瘤也是女性多于男性。肺癌、食管癌、肝癌、胃癌、鼻咽癌和结肠癌等则以男性为多见。性别上的这种差异,其原因除一部分与女性激素有关外,主要是可能与男女的性染色体的不同和某一性别较多地接受某种致癌因子的作用有关。
年龄在肿瘤的发病上也有一定的意义。一般来说,肿瘤的发生随年龄的增大而增加,这可用体细胞突变累积来解释。而多见于幼儿和儿童的肿瘤,常与遗传性的基因损害有关,如视网膜母细胞瘤、肾母细胞瘤和神经母细胞瘤等。
(3)种族和地理因素:某些肿瘤在不同种族或地区中的发生率有相当大的差别,如欧美国家的乳腺癌年死亡率是日本的4~5倍,而日本的胃癌年死亡率比美国高7倍。在我国广东、四川和香港、新加坡等地广东人中,鼻咽癌相当常见而且发病年龄较轻。这说明肿瘤与种族有一定的关系。但是也有移民材料说明移居美国的华侨和日侨中,胃癌的发生率在第三代已有明显的下降。因此地理和生活习惯可能也起到一定的作用。
综上所述,随着分子生物学的发展,近年来对于肿瘤的病因与发病机制的研究有了很大的进展。但是肿瘤的发生发展是异常复杂的,目前了解的只是一角,还有许多未知的领域。但以下几点是迄今比较肯定的:①肿瘤从遗传学上的角度上来说是一种基因病。②肿瘤的形成是瘤细胞单克隆性扩增的结果。③环境的和遗传的致癌因素引起的细胞遗传物质(DNA)改变的主要靶基因是原癌基因和和肿瘤抑制基因。原癌基因的激活和(或)肿瘤抑制基因的失活可导致细胞的恶性转化。④肿瘤的发生不只是单个基因突变的结果,而是一个长期的、分阶段的、多种基因突变积累的过程。⑤机体的免疫监视体系在防止肿瘤发生上起重要作用,肿瘤的发生是免疫监视功能丧失的结果。
第八章 心血管疾病
目前我国经济建设日益发展,人民生活水平不断提高,城市和农村的疾病谱正在变化,传染病已减少,人均寿命在延长,心血管疾病特别是高血压、脑卒中及冠心病的发病和死亡率较30年前有明显升高。1985年我国城市和农村的前三位死因均为心脏病、脑血管病和恶性肿瘤。由于我国城乡肺心病死亡率均较高,经调整后,前三位死因为脑血管病、恶性肿瘤及呼吸系病,心脏病居第4位。1985年我国城市冠心病标准化死亡率为43.7/10万,与法国接近;农村为20.5/10万,与日本接近,但总的说明显低于发达国家。据北京城区1984年调查资料,脑卒中死亡率为男性83.5/10万,女性69.9/10万。若将心脏病与脑血管病合在一起,则居各类主要死亡原因之首。由此可见,心血管疾病的严重性。
第一节 动脉粥样硬化
动脉粥样硬化(atherosclerosis)是严重危害人类健康的常见病。近年来,本病的发病率在我国有明显增加的趋势。据尸检结果,在40~49岁的人群中,冠状动脉和主动脉粥样硬化病变的检出率分别为58.36%和88.31%,并随着年龄的增长而逐渐增加。
动脉硬化(arteriosclerosis)一般是指一组动脉的硬化性疾病,包括:动脉粥样硬化、Mönckeberg动脉中膜钙化和细动脉硬化。
一、动脉粥样硬化的危险因素
1.高脂血症众所周知,高脂血症(hyperlipemia)是动脉粥样硬化的重要危险因素。高脂血症实际上也可认为是高脂蛋白血症,一般以成人空腹12~14小时血甘油三酯超过160mg/dl(1.81mmol/L),胆固醇超过260mg/dl(6.76mmol/L)为高脂血症。大量流行病学调查证明,血浆低密度脂蛋白(LDL)、极低密度脂蛋白(VLDL)水平持续升高与动脉粥样硬化的发病率呈正相关。越来越多的资料表明,LDL必须以某种方式修饰后(如氧化修饰)才致病。近年来,许多学者开始改用纯的脂蛋白颗粒进行研究,例如脂蛋白A(Lp(a))是一种混合颗粒,这种颗粒含有特别多的碳水化合物,它通过影响脂质代谢参与动脉粥样硬化的发生。目前我国人仍多以碳水化合物为主食,高碳水化合物膳食易发生高甘油三酯血症。已知高甘油三酯是本病的独立危险因素。相反,高密度脂蛋白(HDL)可通过胆固醇逆向转运机制清除动脉壁的胆固醇,将其转运至肝代谢并排出体外。此外,HDL有抗氧化作用,防止LDL氧化,并可通过竞争性抑制阻抑LDL与内皮细胞的受体结合而减少其摄取,因此,HDL有抗动脉粥样硬化作用。
2.高血压据统计,高血压患者与同年龄组、同性别的人相比较,其动脉粥样硬化发病较早,病变较重。高血压时血流对血管壁的剪应力(shear stress,即血流冲击力)较高,同时,高血压可引起内皮损伤和(或)功能障碍,从而造成血管张力增高、脂蛋白渗入内膜、单核细胞粘附并迁入内膜、血小板粘附及中膜平滑肌细胞(SMC)迁入内膜等一系列变化,促进动脉粥样硬化发生。另方面,高血压时有脂质和胰岛素代谢异常。有报道认为,高血压患者脂质异常较血压正常者多见;高血压患者有高胰岛素血症及胰岛素抗性(患者对胰岛素不敏感,给予胰岛素患者的血糖降低不明显;给患者口服葡萄糖刺激后,胰岛素释放反应显著增高)。这些均可促进动脉粥样硬化发生。
3.吸烟大量吸烟可使血液中LDL易于氧化,并导致血内一氧化碳浓度升高,从而造成血管内皮缺氧性损伤;烟内含有一种糖蛋白,可激活凝血因子Ⅻ及某种致突变物质,后者可引起血管壁SMC增生。吸烟可使血小板聚集功能增强及血液中儿茶酚胺浓度升高,但使不饱和脂肪酸及HDL水平降低。这些均有助于动脉粥样硬化的发生。
4.性别女性的血浆HDL水平高于男性,而LDL水平却较男性为低。女性在绝经期前动脉粥样硬化的发病率低于同龄组男性,但在绝经期后这种性别差异即告消失,这是由于雌激素能影响脂类代谢,降低血浆胆固醇水平的缘故。
5.糖尿病及高胰岛素血症糖尿病患者的血液HDL水平较低,而且由于高血糖可致LDL糖基化及高甘油三酯血症,后者可产生小而紧密的LDL颗粒,这种LDL较易氧化。这些修饰的LDL可促进血液单核细胞迁入内膜及转变为泡沫细胞。另外,大量调查资料证明,高胰岛素血症(hyperinsulinemia)与动脉粥样硬化的发生密切相关。胰岛素水平越高,冠状动脉心脏病(冠心病)的发病率及死亡率越高,反之,冠心病的发病率及死亡率较低。高胰岛素水平可促进动脉壁SMC增生,而且胰岛素水平与血HDL含量呈负相关。
6.贵传因素冠心病的家族聚集现象提示贵传因素是本病的危险因素。家族性高胆固醇血症(familial hypercholesterolemia,FH)患者由于细胞的LDL受体基因突变以致其功能缺陷,导致血浆LDL水平极度升高。已知,至少有20种贵传性脂蛋白疾病,除FH外,如家族性高乳糜微粒血症(familial hyperchylomicronemia)(血浆乳糜微粒增高)、家族性脂蛋白脂酶缺乏(familial lipoprotein lipase deficiency)(血浆乳糜微粒增高)、家族性高甘油三酯血症(familial hypertriglyceridemia)(血浆VLDL、乳糜微粒增高、HDL降低)及家族性联合高脂血症等。
二、动脉粥样硬化发生机制学说
动脉粥样硬化的发病机制至今尚未完全明了,主要学说有:
1.脂源性学说此说基于高脂血症与本病的因果关系。实险研究也证明,给动物喂饲富含胆固醇和脂肪的饮食可引起与人类动脉粥样硬化相似的血管病变。高脂血症可引起内皮细胞损伤和灶状脱落,导致血管壁通透性升高,血浆脂蛋白得以进入内膜,其后引起巨噬细胞的清除反应和血管壁SMC增生,并形成斑块。Anitschkow(1925)的浸润学说、Rössle(1943)的渗入学说,以及Doerr(1963)的灌注学说都是在这样的事实基础上建立的。
2.致突变学说此学说为EP Benditt和JM Benditt(1973)所提出,认为动脉粥样硬化斑块内的平滑肌细胞为单克隆性,即由一个突变的SMC产生子代细胞,迁移入内膜,分裂增生而形成斑块,犹如平滑肌瘤一般。此起突变的原因可能是化学致突变物或病毒,其根据是,若女性的二倍体强胞核中X染色体的任一个基因是杂合子,机体将由两种不同等位基因型的细胞混合组成(镶嵌性)。目前以6-磷酸葡萄糖脱氢酶(G-6-PD)作为检测这两个等位基因的标记物。G-6-PD有两上异构体(A及B)。若增生病变来自镶嵌个体的单个细胞,则这种病变与正常组织含有两个表型相反,仅含有一个表型的G-6-PD。Benditt等在检查杂合子黑人妇女的正常主动脉及斑块中发现,斑块由产生一种表型的G-6-PD的SMC组成,而正常动脉壁则由两种表型的G-6-PD的SMC混合组成。因此认为这些病变是单克隆来源。
3.损伤应答学说此说为Ross(1976)所提出,1986年又加以修改,认为动脉粥样硬化斑块形成至少有两个途径:①各种原因(机械性、LDL、高半胱氨酸、免疫性、毒素、病毒等)引起内皮损伤,使之分泌生长因子(growth factor,GF),并吸引单核细胞粘附于内皮。单核细胞迁移入内皮下间隙,摄取脂质,形成脂纹,并释放血小板源性生长因子(PDGF)样生长因子。脂纹可直接演变为纤维斑块,或由于内皮细胞脱落而引起血小板粘附。这样,血小板、巨噬细胞及内皮细胞均可产生生长因子,刺激中膜SMC增生。增生病灶内的SMC也可分泌PDGF样生长因子。②内皮细胞受损,但尚完整,内皮细胞更新增加,并产生生长因子,从而刺激中膜SMC迁移进入内膜,SMC及受损内皮细胞均可产生PDGF样生长因子,这种相互作用导致纤维斑块形成,并继续发展。
动脉粥样硬化的炎症学说:损伤应答学说实际上也是一种炎症观点。近年来,随着研究工作的不断深入,动脉粥样硬化发生的炎症学说又重新被强调。
4.受体缺失学说Brown和Goldstein(1973)首先发现人纤维母细胞有LDL受体。已知,该受体广泛分布于肝、动脉壁等全身各种组织细胞膜表面。血浆LDL与LDL受体结合后,聚集成簇,被内吞入细胞,并与溶酶体融合。在溶酶体酶的作用下,LDL中的apo B100被水解为氨基酸,胆固醇酯被水解为游离胆固醇及脂肪酸,前者通过以下途径调节细胞的胆固醇代谢:①抑制内质网的HMG CoA还原酶而抑制细胞本身胆固醇合成;②在转录水平上抑制细胞LDL受体蛋白质的合成;③激活内质网脂酰CoA胆固醇脂酰转移酶(ACAT)活性,使游离胆固醇酯化而储存于胞浆内。LDL被细胞摄取的量取决于细胞膜上受体的多少,若LDL受体数目过少,则导致细胞从循环中清除LDL减少,从而使血浆LDL升高。家族性高胆固醇血症是常染色体显性遗传病,患者由于细胞表面LDL受体功能缺陷而导致血浆LDL水平极度升高。患者多在早年发生冠心病而死亡。
三、病理变化
动脉壁的年龄变化:据国内研究,早在3个月胎龄时即见到主动脉内弹力膜分层,中膜浅层SMC空过弹力膜窗孔进入内膜,其后SMC增生,产生胶原、弹性纤维及蛋白多糖。内膜随着年龄增长而逐渐增厚。此外,在动脉杈或分支开口处常见小块白色增厚区,称为内膜垫(intimal cushion)。内膜垫由SMC、胶原纤维及蛋白多糖组成,可能是对血流剪应力的反应。据病理普查结果表明,动脉粥样硬化病变的发生与年龄的关系十分密切。动脉杈、分支开口及血管弯曲的凸面为病变的好发部位。
1.脂纹脂纹(fatty streak)是动脉粥样硬化的早期病变。据尸检普查,9岁以下儿童的主动脉脂纹检出率为11.5%,10~19岁为48.96%。肉眼观,主动脉的脂纹常见于其后壁及分支开口处,为帽针头大小斑点及宽约1~2mm、长短不一的黄色条纹,不隆起或稍微隆起于内膜表面。
脂纹的形成多先有高脂血症,高脂血症或其它有害因子可造成内皮损伤,使其表面糖萼变薄,内皮细胞间间隙增宽。LDL与内皮细胞的高亲和性受体结合而被摄取,通过胞浆,进入内皮下间隙,并被内皮细胞及SMC释放的氧自由基氧化修饰,产生氧化LDL(OX-LDL)及氧化Lp(a)[OX-Lp(a)]。
在动脉分杈、分支开口处以及变曲动脉的凸面的血流剪应力减低,并可出现涡流,这使单核细胞易离开轴流与内皮接触。已知内皮细胞能分泌几种粘附分子,例如细胞间粘附分子(intercellular adhesion molecule-1,ICAM-1)及血管粘附分子(vascular adhesion molecule-1,VCAM-1)。ICAM-1可与白细胞表面的受体β2整合素(β2integrin,包括LFA-1及MAC-1)结合,VCAM-1可与白细胞的受体(VLA-4)结合,从而使单核细胞粘附于内皮表面。
单核细胞迁入内皮下间隙受多种因素影响。其中最重要的是SMC分泌的单核细胞趋化蛋白1(monocyte chemotactic protein 1,MCP-1),对单核细胞有很强的趋化活性。此外,动脉壁细胞产生的生长因子(如PDGF)及OX-LDL等对单核细胞亦有趋化活性。迁入内皮下间隙的单核细胞被激活并分化成巨噬细胞。
OX-LDL、OX-Lp(a)可与巨噬细胞表面的清道夫受体结合而被摄取。这些受体对胆固醇无下调作用,因而被巨噬细胞摄取的脂质愈来愈多,直至形成泡沫细胞(foam cell)(图8-1)。
图8-1 单核细胞迁入内膜及泡沫细胞形成模式图
LDL渗入内皮下间隙(SES),被氧自由基氧化修饰;MCP-1释放,单核细胞(MC)迁入内膜,OX-LDL与巨噬细胞表面的清道夫受体结合而被摄取,泡沫细胞形成(EC:内皮细胞,SMC:平滑肌细胞)(仿Schwartz)
大量泡沫细胞聚集即形成脂纹,内皮隆起及变形。电镜下,巨噬细胞源性泡沫细胞表面富有突起,形成丝状伪足;胞浆内含有大量较小的脂质空泡和溶酶体,有时还见到胆固醇结晶;核卵圆或略呈肾形,异染色质常呈块状紧靠核膜,偶见1~2个核仁。内皮细胞、巨噬细胞及SMC均可分泌生长因子〔PDGF、纤维母细胞生长因子(FGF)、表皮生长因子(EGF)等〕,在其作用下,原已存在于内膜的SMC增生;中膜SMC发生表型转变(phenotypic modulation),即由收缩型(胞浆内含大量肌丝及致密体)转变为合成型(含大量粗面内质网、核蛋白体及线粒体);同时,SMC穿过内弹力板窗孔迁移入内皮下间隙并增生。SMC表面有LDL受体,可结合、摄取LDL及VLDL而成为泡沫细胞(肌源性泡沫细胞)。电镜下,肌源性泡沫细胞多呈长形,或有突起,多少保持SMC的特点,胞浆内可见肌丝和致密体,脂质空泡多少不定,一般稍大,有时能见到基底膜(图8-2)。
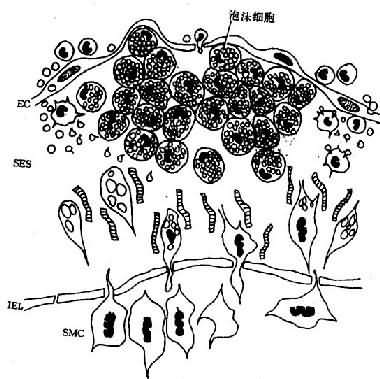
图8-2 脂纹组织结构模式图
内皮下间隙(SES)大量巨噬细胞源性泡沫细胞聚集,中膜SMC穿过内弹性膜(IEL)窗孔迁入内膜,增生并摄取脂质,内膜隆起及变形EC:内皮细胞病变的进展:已知OX-LDL具有细胞毒性,当大量OX-LDL被巨噬细胞摄取,超过了其清除能力时可引起泡沫细胞的坏死,导致细胞外脂质核心形成。加之,SMC大量增生,穿插于巨噬细胞源性泡沫细胞之间,产生胶原、弹性纤维及蛋白多糖,使病变演变为纤维斑块(图8-3)。
2.纤维斑块肉眼观,纤维斑块(fibrous plaque)为隆起于内膜表面的灰黄色斑块。随着斑块表层的胶原纤维不断增加及玻璃样变,脂质被埋于深层,斑块乃逐渐变为瓷白色。镜检下,斑块表面为一层纤维帽,乃由多量SMC及大量细胞外基质(包括胶原、弹性纤维、蛋白聚糖及细胞外脂质)组成。纤维帽之下有不等量的增生的SMC、巨噬细胞及两种泡沫细胞,以及细胞外脂质及基质(图8-4,图8-5)。
由于OX-LDL的细胞毒性作用,以及很可能内皮细胞及SMC产生的氧自由基的作用,可引起斑块内细胞损伤及坏死。比较脆弱的巨噬细胞源性泡沫细胞坏死后,其胞浆内的脂质被释放出来,成为富含胆固醇酯的脂质池。泡沫细胞坏死崩解,释放出许多溶酶体酶,促进其它细胞坏死崩解。随着这些病理过程的发展,纤维斑块逐渐演变为粥样斑块。
3.粥样斑块粥样斑块(atheromatous plaque)亦称粥瘤(atheroma)。肉眼观,为明显隆起于内膜表面的灰黄色斑块。切面,表层的纤维帽为瓷白色,深部为多量黄色粥糜样物质(由脂质和坏死崩解物质混合而成)(图8-6)。镜检下,纤维帽趋于老化,胶原纤维陷于玻璃样变,SMC被分散埋藏在细胞外基质之中。深部为大量无定形坏死物质,其内富含细胞外脂质,并见胆固醇结晶(石蜡切片上为针状空隙)、钙化等(图8-7)。底部和边缘可有肉芽组织增生,外周可见少许泡沫细胞和淋巴细胞浸润。病变严重者中膜SMC呈不同程度萎缩,中膜变薄。外膜可见新生毛细血管、不同程度的结缔组织增生及淋巴细胞、浆细胞浸润。一些学者认为,这种外膜炎症(慢性主动脉周围炎)可能是对粥瘤中的类蜡质(ceroid 一种含高度不饱和脂肪酸的黄色腊样物质)成分的一种自身免疫反应。
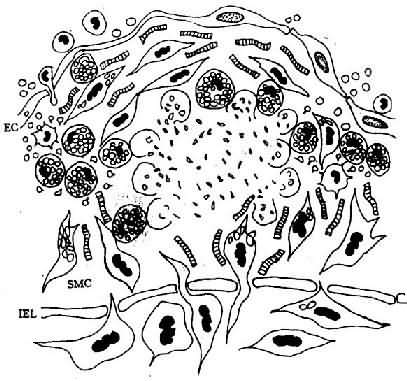
图8-3 脂纹的进展
泡沫细胞坏死及细胞外脂质核心形成,SMC继续增生,产生胶原、弹性纤维及蛋白多糖,使病变演变为纤维斑块

图8-4 发展中的纤维粥样斑块
斑块表层为脂纹,含有泡沫细胞和增生的平滑肌细胞;深层有大量胆固醇(针状结晶)析出和泡沫细胞散在。男性,25岁,主动脉。
(阜外医院病理科供图)
图8-5 发展中的纤维粥样斑块
图8-4的斑块深层。高倍镜下可见大量胆固醇结晶、泡沫细胞和增生的平滑肌细胞

图8-6 主动脉粥样硬化
主动脉内膜面见粥样斑块形成
4.复合病变
(1)斑块内出血:在粥样斑块的边缘常见到许多薄壁的新生血管。在血流剪应力作用下,这些薄壁血管常易破裂出血,可形成血肿,使斑块更加隆起,其后血肿被机化。
(2)斑块破裂:为最危险的并发症,斑块破裂常形成溃疡(粥瘤性溃疡)及并发血栓形成;坏死性粥样物质可排入血流而造成胆固醇栓塞。斑块破裂常见于腹主动脉下端、髂动脉和股动脉。富含软的细胞外脂质的斑块,特别是脂质池偏位时,容易破裂。斑块外周部分纤维帽最薄,含胶原、氨基葡聚糖及SMC较少,细胞外脂质较多,抗张强度较差,该处巨噬细胞源性泡沫细胞浸润也最多。因此,破裂往往发生在纤维帽的外周。
(3)血栓形成:表浅的或由于斑块破裂造成较深的内膜损伤,均可使胶原暴露,通过von Willebrand因子的介导,引起血小板的聚集而形成血栓,可引起器官动脉阻塞而导致梗死(如脑梗死、心肌梗死)。冠状动脉血栓的机化及再通可在一定程度上恢复该支动脉的血流,有助于保持梗死灶周围心肌的存活。
(4)钙化:多见于老年患者,钙盐可沉积于坏死灶及纤维帽内,动脉壁因而变硬、变脆。钙化灶可进而发生骨化。

图8-7 主动脉粥样硬化
内膜中形成粥瘤,其中为一些坏死物质及胆固醇结晶,表面内膜呈纤维性增厚
(5)动脉瘤形成:严重的粥样斑块底部的中膜SMC可发生不同程度的萎缩,以致逐渐不能承受血管内压力(张力)的作用而扩张,形成动脉瘤(aneurysm)。另外,血流可从粥瘤性溃疡处侵入主动脉中膜,或中膜内血管破裂出血,均可造成中膜撕裂,形成夹层动脉瘤(aneurysma dissecans)。
四、重要器官的动脉粥样硬化
(一)主动脉粥样硬化
病变多发生于主动脉后壁和其分支开口处。腹主动脉病变最严重,其次是降主动脉和主动脉弓,再次是升主动脉(图8-8)。病变严重者,斑块破裂,形成粥瘤性溃疡,其表面可有附壁血栓形成。有的病例因中膜SMC萎缩,弹力板断裂,局部管壁变薄弱,在血压的作用下管壁向外膨出而形成主动脉瘤。这种动脉瘤主要见于腹主动脉。偶见动脉瘤破裂,发生致命性大出血。有时可发生夹层动脉瘤。有的病例主动脉根部内膜病变严重,累及主动脉瓣,使瓣膜增厚、变硬,甚至钙化,形成主动脉瓣膜病。
图8-8 主动脉粥样硬化
在主动脉内膜面上见隆起的粥样斑块并有溃疡形成
(二)冠状动脉粥样硬化
详见本章第十节。
(三)脑动脉粥样硬化
脑动脉粥样硬化发生较迟,一般在40岁以后才出现斑块。病变以Willis环和大脑中动脉最显著(图8-9)。据近年来报道,颈内动脉起始部及颅内部的粥样硬化病变相当常见,可有不同程度的管腔狭窄、斑块内出血、溃疡及附壁血栓形成。
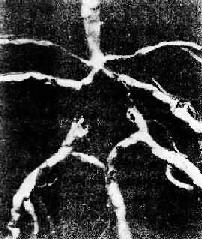
图8-9 脑底动脉Willis环及其分支粥样硬化
本病时由于脑动脉管腔狭窄,脑组织因长期供血不足而发生萎缩。大脑皮质变薄,脑回变窄,脑沟变宽、加深,重量减轻。严重者常有智力减退,甚至痴呆。
严重的脑动脉粥样硬化使管腔高度狭窄,常继发血栓形成而导致管腔阻塞,脑组织缺血而发生梗死(脑软化)。脑软化多见于颞叶、内囊、尾状核、豆状核和丘脑等部位。镜检下,脑软化灶早期,组织变疏松,神经细胞变性、坏死,数量减少,周围有少量炎性细胞浸润。由小胶质细胞转变来的巨噬细胞摄取坏死组织崩解产生的脂质,使胞体增大,胞浆呈泡沫状。小软化灶可被吸收,由胶质细胞增生修复。较大的软化灶周围由增生的胶质纤维和胶原纤维围绕,坏死组织液化吸收形成囊腔。严重脑梗死可引起病人失语、偏瘫,甚至死亡。发生在延髓的软化灶可引起呼吸、循环中枢麻痹。脑动脉粥样硬化病变可形成小动脉瘤,当血压突然升高时可破裂出血。
(四)肾动脉粥样硬化
据统计,80%肾动脉粥样硬化性狭窄见于肾动脉开口处或主干近侧端,多为偏心的纤维斑块。严重者可导致肾动脉高度狭窄,甚或因并发血栓形成而完全阻塞。前者引起肾血管性高血压,后者引起受累动脉供血区域的梗死,梗死灶机化后形成较大块的凹陷瘢痕。多个瘢痕使肾缩小,称为动脉粥样硬化性固缩肾。
(五)四肢动脉粥样硬化
下肢动脉粥样硬化较上肢为常见,且较严重。股浅动脉在内收肌腱裂孔水平处最常发生阻塞,可能是由于动脉在该处易受大内收肌硬的腱弓反复机械作用所致。四肢动脉吻合支较丰富,较小的动脉管腔逐渐狭窄以至闭塞时,一般不发生严重后果。当较大动脉管腔明显狭窄时,可因肢体缺血在行走时出现跛行症状。当动脉管腔严重狭窄,继发血栓形成而侧支循环又不能代偿时,可发生供血局部的缺血性坏死(梗死),甚至发展为坏疽(图8-10)。
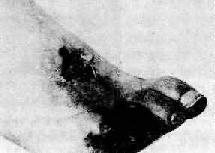
图8-10 右足趾坏疽
第二节 高血压
高血压(hypertension)为人类最常见的疾病之一,据流行病学调查,估计我国现有高血压患者约5000万人,每年新发病例约120万人。广义而言,体循环长期持续的不正常的血压升高称为高血压。
正常人的血压在不同的生理状况下有一定的波动幅度。收缩压和舒张压均随年龄的增长而升高,但是,舒张压升高不明显。因此,舒张压升高是判断高血压的重要依据。据世界卫生组织(WHO)建议,高血压的诊断标准如下:
1.正常成人血压 收缩压为18.6kPa(140mmHg)或以下,舒张压在12kPa(90mmHg)或以下。
2.成年人高血压 收缩压为21.3kPa(160mmHg)或以上和(或)舒张压在12.6kPa(95mmHg)或以上。
3.临界性高血压 收缩压高于18.6kPa而低于21.3kPa,舒张压高于12kPa而低于12.6kPa。
高血压可分为特发性高血压(essential hypertension)和继发性高血压(secondary hypertension)。前者又称原发性高血压(primary hypertension),后者又称为症状性高血压(symptomatic hypertension)。前者原因尚未完全明了,后者由某些疾病引起,如慢性肾小球肾炎、肾动脉狭窄、肾上腺和垂体的肿瘤等。本节仅叙述原发性高血压,继发性高血压将在以后各章节有关疾病中述及。
原发性高血压比较常见,多发生于中年(35~40岁)以后,男性与女性的患病率无明显差别。
一、病因和发病机制
原发性高血压的病因和发病机制尚未完全明了,近年来,随着生理学、遗传学、神经内分泌学及分子生物学的发展,以及自发性高血压大鼠模型的建立,许多研究已达到分子水平。目前,各种学说中以Page的镶嵌学说(mosaic theory)比较全面,认为高血压并非由单一因素引起,而是由彼此之间相互影响的多种因素造成。
1.遗传因素约75%的原发性高血压患者具有遗传素质(genetic predisposition),同一家族中高血压患者常集中出现。据信原发性高血压是多基因遗传病。据报道,高血压患者及有高血压家族史而血压正常者有跨膜电解质转运紊乱,其血清中有一种激素样物质,可抑制Na+/K+-ATP酶活性,以致钠钾泵功能降低,导致细胞内Na+、Ca2+浓度增加,动脉壁SMC收缩加强,肾上腺素能受体(adrenergic receptor)密度增加,血管反应性加强。这些都有助于动脉血压升高。近来研究发现,血管紧张素(AGT)基因可能有15种缺陷,正常血压的人偶见缺陷,而高血压患者在AGT基因上的3个特定部位均有相同的变异。患高血压的兄弟或姐妹可获得父母的AGT基因的同一拷贝。有这种遗传缺隐的高血压患者,其血浆血管紧张素原水平高于对照组。
2.膳食电解质一般而言,日均摄盐量高的人群,其血压升高百分率或平均血压高于摄盐量低者。WHO在预防高血压措施中建议每人每日摄盐量应控制在5g以下。一项由32个国家参加共53个中心关于电解质与血压关系的研究结果表明,中国人群尿钠平均值为206mmol/24h,比其它中心高43mmol/24h,尿钠/钾比达6.7,是其它中心的2倍多。尿钠最高的是天津(242mmol/24h)。这与中国膳食的高钠、低钾有关。钾能促进排钠,吃大量蔬菜可增加钾摄入量,有可能保护动脉不受钠的不良作用影响。钙可减轻钠的升压作用,我国膳食普遍低钙,可能加重钠/钾对血压的作用。增加膳食钙摄量的干预研究表明,钙的增加使有些患者血压降低。
3.社会心理应激据调查表明,社会心理应激与高血压发病有密切关系。应激性生活事件包括:父母早亡、失恋、丧偶、家庭成员车祸死亡、病残、家庭破裂、经济政治冲击等。遭受生活事件刺激者高血压患病率比对照组高。据认为,社会心理应激可改变体内激素平衡,从而影响所有代谢过程。
4.肾因素肾髓质间质细胞分泌抗高血压脂质如前列腺素、抗高血压中性肾髓质脂等分泌失调,排钠功能障碍均可能与高血压发病有关。
5.神经内分泌因素一般认为,细动脉的交感神经纤维兴奋性增强是本病发病的重要神经因素。但是,交感神经节后纤维有两类:①缩血管纤维,递质为神经肽Y(neuropeptide Y,NPY)及去甲肾上腺素;②扩血管纤维,递质为降钙素基因相关肽(calcitonin gene related peptide,CGRP)及P物质。这两种纤维功能失衡,即前者功能强于后者时,才引起血压升高。近年来,中枢神经递质和神经肽,以及各种调节肽与高血压的关系已成为十分活跃的研究领域。据报道,CGRP可能抑制大鼠下丘脑去甲肾上腺素的释放,在外周它可能抑制肾上腺神经受刺激时去甲肾上腺素的释放。有报道,从哺乳动物心脏和脑中分离出利钠肽(A、B及C型),启示了人体内有一个利钠肽家族。近来在局部肾素-血管紧张素系统(RAS)的研究取得了新进展。将小鼠肾素基因(Ren-2基因)经微注射装置注入大鼠卵细胞,形成了转基因大鼠种系TGR(mREN2)27,这种动物血压极高。用Northern印迹杂交法证明,Ren-2转基因表达在肾上腺、血管、胃肠及脑,并可表达于胸腺、生殖系统和肾。由于其表达于血管壁,可能使血管的血管紧张素形成增加,从而发生高血压和血管SMC肥大。
二、类型和病理变化
(一)良性高血压
良必高血压(benign hypertension)又称缓进型高血压。早期多无症状,往往是偶然发现。开始表现为全身细动脉和小动脉痉挛,呈间断性,血压亦处于波动状态,其后血压呈持续性升高。出现心血管并发症者,如冠状动脉粥样硬化,促进疾病的进展。晚期可因心力衰竭、心肌梗死或脑出血致死。因肾功能衰竭致死者少见。
1.动脉系统病变
(1)细动脉:是指中膜仅有1~2层SMC的细动脉(arteriole)及直径约1mm 及以下的最小动脉。常累及腹腔器官、视网膜及肾上腺包膜的细动脉,最严重的是肾脏入球动脉。由于细动脉反复痉挛,血管内压持续升高,内皮细胞虽可通过其细胞骨架的适应性来加强适应,但仍不能承受血管内压力升高的作用而被分开,内皮细胞间间隙扩大,血浆蛋白(含免疫球蛋白及纤维蛋白原)渗入内皮下间隙。局部区域中膜SMC可发生坏死,溶酶体酶释出,并可引起局部性蛋白溶解,以致该处管壁通透性升高。血浆蛋白的渗入连同由未坏死SMC产生的修复性胶原纤维及蛋白多糖使细动脉壁细胞愈来愈减少而陷于玻璃样变,形成细动脉硬化(arteriolosclerosis)。镜检下,细动脉内皮与中膜SMC之间有玻璃样物质沉积,其内的胶原纤维亦陷于均质化。随着疾病的发展,细动脉管壁愈来愈增厚、变硬,管腔狭窄,甚至可使管腔闭塞。
(2)肌型器官动脉:主要累及冠状动脉、脑动脉及肾动脉(弓形及小叶间动脉常被累及)。表现为中膜SMC肥大和增生,中膜内胶原、弹性纤维及蛋白多糖增加,使中膜增厚。内膜亦有血浆蛋白渗入,SMC增生,产生胶原和弹性纤维,内弹力膜分裂,管腔可有某种程度狭窄。
2.心脏的病变 主要为左心室肥大,这是对持续性血压升高,心肌工作负荷增加的一种适应性反应。在心脏处于代偿期时,肥大的心脏心腔不扩张,甚至略微缩小,称为向心性肥大(concentric hypertrophy)。心脏重量增加,一般达400g以上,甚至可增重1倍。肉眼观,左心室壁增厚,可达1.5~2cm;左心室乳头肌和肉柱明显增粗(图8-11)。镜检下,肥大的心肌细胞变粗,变长,并有较多分支。细胞核较长、较大(可形成多倍体)。由于不断增大的心肌细胞与毛细血管供养之间的不相适应,加上高血压性血管病,以及并发动脉粥样硬化所致的血供不足,便导致心肌收缩力降低,逐渐出现心腔扩张,称为离心性肥大(eccentric hypertrophy)。严重者可发生心力衰竭。
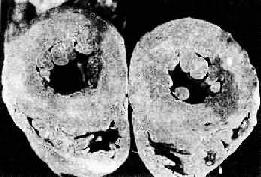
图8-11 高血压病时左心室向心性肥大(心脏横断面)
左心室壁增厚,乳头肌显著增粗
3.肾的病变 表现为原发性颗粒性固缩肾,为双侧对称性、弥漫性病变。
肉眼观,肾体积缩小,质地变硬,重量减轻,一侧肾重量一般小于100g(正常成年人一侧肾重约为150g)。表面布满无数均匀的红色细颗粒。切面,肾皮质变薄,一般在2mm左右(正常厚3~5mm)。髓质变化不明显,但肾盂和肾周围脂肪组织明显增生。
镜检下,肾细动脉硬化明显,小叶间动脉及弓形动脉内膜增厚。依病程而有多少不等的入球动脉及肾小球发生玻璃样变(图8-12)。附近的肾小管由于缺血而萎缩、消失,间质结缔组织增生及淋巴细胞浸润。该处由于肾实质萎缩和结缔组织收缩而形成凹陷的固缩病灶,周围健存的肾小球发生代偿性肥大,所属肾小管亦呈代偿性扩张,使局部肾组织向表面隆起,形成肉眼所见的无数红色细颗粒(由于该处血供良好而呈红色)。
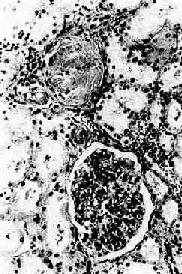
图8-12 高血压病之肾
肾小球入球小动脉管壁玻璃样变性,肾小球纤维化,玻璃样变
临床上,可多年不出现肾功能障碍。晚期由于病变的肾单位越来越多,肾血流量逐渐减少,肾小球滤过率逐渐降低。患者可发生水肿、出现蛋白尿及管型。严重者可出现尿毒症的临床表现。
4.脑的病变 高血压时,由于脑内细动脉的痉挛和病变,患者可出现不同程度的高血压脑病(hypertensive encephalopathy)症状,如头痛、头晕、眼花等,甚至出现高血压危象。患者有明显的中枢神经症状,如意识模糊、剧烈头痛、恶心、呕吐、视力障碍及癫痫发作等。
(1)脑动脉病变:严重的病例细动脉和小动脉管壁可发生纤维素样坏死,可并发血栓形成及微动脉瘤(microaneurysm)。后者好发于壳核、丘脑、脑桥、小脑和大脑,这些部位也是高血压性脑出血及脑梗死发生率最高之处。
(2)脑软化:由于细动脉、小动脉病变造成其所供养区域脑组织缺血的结果,脑组织内可出现多数小软化灶,即微梗死灶(microinfarct)。镜检下,梗死灶内脑组织坏死液化,形成染色较浅、质地疏松的筛网状病灶。灶内可见坏死的细胞碎屑,周围有胶质细胞增生及少量炎性细胞浸润。最后,坏死组织被吸收,由胶质瘢痕修复。由于软化灶较小,一般不引起严重后果。
(3)脑出血:是高血压最严重的且往往是致命性的并发症。多为大出血灶,常发生于基底节、内囊,其次为大脑白质、脑桥和小脑。出血区域的脑组织完全被破坏,形成囊腔状,其内充满坏死的脑组织和凝血块。有时出血范围甚大,可破入侧脑室(图8-13)。引起脑出血的原因一方面由于细、小动脉的病变,另一方面,脑出血多发生于基底节区域(尤以豆状核最多见),供养该区的豆纹动脉从大脑中动脉呈直角分出,直接受到大脑中动脉压力较高的血流冲击,易使已有病变的豆纹动脉破裂出血。此外,血压突然升高(如情绪激动时)亦易使病变的动脉破裂出血。临床上,患者常骤然发生昏迷、呼吸加深和脉搏加快。严重者可发生陈-施(Cheyne-Stokes)呼吸、瞳孔反射及角膜反射消失、肢体弛缓、肌腱反射消失、大小便失禁等症状。出血灶扩展至内囊时,引起对侧肢体偏瘫及感觉消失。出血灶破入侧脑室时,患者发生昏迷,常导致死亡。左侧脑出血常引起失语,脑桥出血可引起同侧面神经麻痹及对侧上下肢瘫痪。
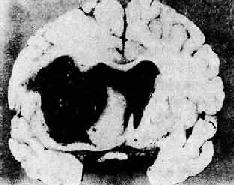
图8-13 高血压病之脑出血
大脑左侧内囊出血并破入侧脑室
5.视网膜的病变 视网膜中央动脉亦常发生硬化。眼底镜检查可见这些血管迂曲,颜色苍白,反光增强,呈银丝样改变。动、静脉交叉处静脉呈受压现象。严重者视乳头发生水肿,视网膜渗出和出血,患者视物模糊。
(二)恶性高血压
恶性高血压(malignant hypertension)又称为急进型高血压,可由良性高血压恶化而来,或起病即为急进性。病理变化主要见于肾和脑。
1.肾的变化 镜检下,可见细动脉坏死(arteriolonecrosis)(纤维素样坏死),坏死累及内膜和中膜,并有血浆成分内渗(insudation),使管壁极度增厚。HE染色切片上,受累血管壁呈嗜伊红性和折光性,免疫组织化学检查证明其中含有纤维蛋白、免疫球蛋白、补体。可见核碎片,但炎性细胞浸润极少见。有时可见到坏死性细动脉炎,在坏死的血管壁及周围有单核细胞及中性粒细胞浸润,但这种动脉炎不累及弓形动脉及叶间动脉。入球动脉坏死常波及肾小球,使肾小球毛细血管丛发生节段性坏死。细动脉坏死常并发血栓形成,可引起出血及微梗死。小动脉的变化颇具特征性,表现为增生性动脉内膜炎(proliferative endarteritis),内膜显著增厚,其内有多数SMC增生,并呈向心性排列,形成层状葱皮样病变。SMC产生大量胶原及蛋白多糖,管腔陷于高度狭窄。然而,这些变化并非恶性高血压所特有,类似的变化亦见于肾移植慢性排斥反应、进行性系统性硬化等。肉眼观,肾表面平滑,可见多数出血点,切面可见多数斑点状微梗死灶。
2.脑的变化 脑的细、小动脉亦可发生同样病变,常引起局部缺血、微梗死和脑出血。
临床上,有严重的高血压,血压值超过30.66/17.3kPa(230/130mmHg)。可发生高血压脑病。常有持续蛋白尿、血尿及管型尿。患者多于一年内因尿毒症、脑出血或心力衰竭致死。
第三节 动脉中膜钙化
动脉中膜钙化(medial calcification)亦称Mбnckeberg中膜硬化,为原因不明的变质性疾病,其特点是动脉中膜发生营养不良性钙化。好发于老年人的下肢大动脉,上肢动脉也可被累及。
【病因和发病机制】
本病的病因和发病机制尚未明了,其发生似乎与年龄变化有很大关系。钙化开始于内弹性膜间隙区,然后扩展至中膜。超威结构研究表明,在该处可见到基质小泡聚集和弹性组织裂隙。基质小泡是一种膜包裹小泡,可能来自SMC的终溶酶体,钙盐首先沉积其中。
【病变】
镜检下,早期,中膜SMC发生玻璃样变,有时可见到小坏死灶,继而有钙盐沉积,钙化灶周围有纤维组织增生,极少或无炎性细胞反应。偶尔在钙化区可出现骨化生现象,化生的骨质中可含有红骨髓。动脉内膜多无明显变化。肉眼观,由于中膜钙化灶呈间隔的环带状,致使整个受累动脉僵硬,并呈鹅喉样外观。
第四节 动脉炎
一、大动脉炎
大动脉炎是指主动脉及其大分支的炎症。主动脉炎包括梅毒性主动脉炎、高安动脉炎、风湿性主动脉炎及类风湿性主动脉炎,其原因有所不同,但其病理变化,无论肉眼观或组织学变化,均非特异性,且都颇为相似,因此不能简单地从肉眼变化及组织学来确定病因。
(一)梅毒性主动脉炎
梅毒是由梅毒螺旋体引起的一种性病。梅毒性主动脉炎见于第三期梅毒,往往在原发感染后十余年才出现。病变特点是主动脉滋养血管周围有淋巴细胞、浆细胞和单核细胞浸润。中膜有粟粒状树胶样肿形成,并可见灶状坏死及弹力板破坏。晚期形成多数小瘢痕。内膜则见高度纤维化。肉眼观,主动脉内膜表面呈典型的树皮样外观。
(二)高安动脉炎
高安动脉炎(Takayasu’s arteritis)亦称特发性主动脉炎(idiopathic aortitis)、无脉症(pulseless disease),多见于东方国家,但世界各地均有发生。在我国,以东北,特别是黑龙江省较为常见。本病多见于青年人,女性多于男性,临床上,依受累血管部位而表现相应症状,并有关节炎、发热、贫血、血沉加速和体重减轻等。
【病因和发病机制】
尚未明了。由于本病主要侵犯大动脉,中膜弹性纤维断裂消失,结缔组织发生粘液变性及纤维素样坏死。有的学者认为本病属胶原病范畴。据报道,患者血清中存在抗主动脉抗体,所以认为本病是一种自身免疫性疾病。
【病变】
病变分布范围较广,可累及整个主动脉及其大分支,包括冠状动脉、锁骨下动脉、颈总动脉、无名动脉、肾动脉、腹腔动脉及肠系膜上动脉。肉眼观,被累主动脉管壁明显增厚、变硬。内膜表面凹凸不平,重者有多数斑块隆起,其间为纵行或星状皱纹(树皮样皱纹)(图8-14)。受累大支管腔明显狭窄,甚至被纤维组织完全阻塞。
图8-14 高安主动脉炎
升主动脉和主动脉弓、胸主动脉上段两个动脉瘤,后者充满血栓物质;肾动脉开口以上的主动脉内膜显著增厚,呈树皮样外观。男性,25岁
镜下,早期,动脉中膜可见粘液样变性及纤维素样坏死灶,继而中膜弹力板有灶状甚至较大范围破坏,伴有网芽组织增生,含有新生毛细血管、不等量结缔组织增生及淋巴细胞浸润,晚期有瘢痕形成。病变最严重处中膜弹力板几乎全被破坏,代之以大量结缔组织,基质中富含蛋白多糖,PAS染成红色,阿新蓝(alcian blue)染成蓝色。内膜显著增厚,其中SMC大量增生并产生大量胶原纤维及蛋白多糖,内弹力膜断裂或消失。外膜大量结缔组织增生,其中胶原纤维常发生玻璃样变;滋养血管壁增厚,其周围有淋巴细胞、浆细胞浸润(图8-15)。

8-15 高安主动脉炎
主动脉中膜弹力板被破坏中断(↑),代之以瘢痕组织,内膜及外膜大量胶原纤维增生,滋养血管壁增厚。弹性纤维染色
本病的变化与梅毒性主动脉炎极为相似,单从形态学上往往很难鉴别,但本病多见于青年人,病变范围较广;梅毒患者有特异的血清反应而本病则否。
【合并症】
动脉瘤形成不太多见,但可为多发性。部分患者有无脉症,多由于头、臂动脉近分支出口处的一段血管管腔被增生的胶原纤维阻塞所引起。肾动脉受累时多有高血压。极少数患者可发生冠状动脉炎,可并发冠状动脉瘤、血栓形成及心肌梗死。
(三)巨细胞性动脉炎
巨细胞性动脉炎(giant cell arteritis)曾被称为颞动脉炎(temporal arteritis)和颅动脉炎(cranial arteritis)。本病几乎仅见于中、老年人,年龄都在50岁以上,女性多于男性,临床上,常有风湿性多肌痛症(polymyalgia rheumatica),可累及颈、肩、上肢、臀部和大腿肌肉。由于颞动脉受累,可出现局限性或弥漫性头痛、头皮触痛、颜面痛等症状。触诊可扪及颞动脉呈结节状增粗。眼动脉被累及时可引起视神经乳头贫血性梗死而使患者失明。全身症状有发热、食欲缺乏和体重减轻。
【病因和发病机制】
病因未明,虽为肉芽肿性炎症,但从病灶中从未分离出微生物,从病变上看,本病似乎是机体对动脉壁某种成分(弹性蛋白)的一种免疫反应。有人报道,有的患者的动脉抗原可引起细胞介导的免疫反应,以及用皮质激素治疗有效等支持这种观点。关于遗传因素,有报道在兄弟姊妹之间的发病率比普通人群高10倍。
【病变】
主要侵犯颅动脉,尤其是颞动脉,但可累及全身的中等大的动脉。肉眼观,被累血管呈结节状增粗,变硬。镜下,早期见动脉中膜SMC变性坏死,伴有弹性纤维破坏及炎性细胞浸润。内弹性膜几乎都发生断裂、破坏,引起肉芽肿性炎症反应,其中含有多核巨细胞、类上皮细胞,以及淋巴细胞、单核细胞浸润(图8-16,图8-17)。内膜常见纤维化,管腔可陷于高度狭窄,可并发血栓形成,并见机化、再通现象。愈复期炎性浸润及巨细胞消失,整个动脉壁陷于纤维化。
图8-16 巨细胞性动脉炎
跟腱旁小动脉中膜被破坏,代之以含许多巨细胞的肉芽组织,管腔高度狭窄HE
图8-17 巨细胞性动脉炎
同上病例。动脉中膜弹力性纤维被破坏。半薄切片,甲苯胺蓝染色
二、坏死性动脉炎
坏死性动脉炎(necrotizing arteritis)包括一些主要因免疫复合物沉积而引起的动脉炎,如结节性多动脉炎、变态反应性肉芽肿性动脉炎、Wegener肉芽肿病及狼疮动脉炎。病变特点:动脉壁多有纤维素样坏死及各种炎性细胞浸润,其中浸润的细胞成分因不同类型而略有差别。
三、血栓闭塞性脉管炎
血栓闭塞性脉管炎(thromboangiitis obliterans)亦称Bürger病。其特点是,多累及中等大的动脉和静脉,特别是下肢的血管,发生复发性炎症和血栓形成,引起循环障碍,出现间歇性跛行(claudicatio intermittens)和肢体坏疽。绝大多数发生于男性,年龄20~40岁。
【病因和发病机制】
病因未明。由于本病几乎仅见于重吸烟者,戒烟可使临床症状缓解,因此认为本病的发生与烟碱有关。有人认为本病是一种独立的血管疾病,其发生是由于对烟碱的过敏反应,加上血液内一氧化碳血红蛋白含量增高,引起血管内膜的炎症,继发血栓形成。
【病变】
早期为小动脉的一些节段发生内膜炎,管壁发生纤维素样坏死,伴有中性粒细胞浸润,可出现多核巨细胞,管腔因并发血栓形成而闭塞。病变逐渐累及较大的动脉段。动脉中膜和外膜亦有炎性细胞浸润,病变常累及伴行的静脉。富含新生毛细血管及炎性细胞的机化血栓据说是本病的特点之一,血栓经机化后可发生再通现象,遗留斑块状内膜瘢痕,中膜弹力板通常无明显破坏。
第五节 动脉瘤
动脉管壁病理性局限性扩张称为动脉瘤(aneurysm)。动脉瘤可依病因、形状和动脉瘤壁的结构进行分类(图8-18)。依病因可分为:动脉粥样硬化性、梅毒性、细菌性、外伤性和先天性动脉瘤,以及夹层动脉瘤。
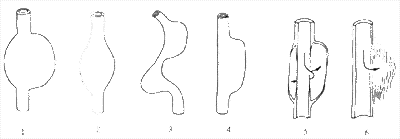
图8-18 动脉瘤的类型
1.囊状动脉瘤;2.梭形动脉瘤;3.蜿蜒状动脉瘤;4.舟状动脉瘤;5.6.假性动脉瘤
一、动脉瘤的形态学类型
1.囊状动脉瘤 被累血管段管壁呈球状扩张,其大者直径可达15~20cm。由于血液流过时形成旋涡,因此,这种动脉瘤常并发血栓形成。
2.梭形动脉瘤 血管壁呈均匀扩张,而又朝一端逐渐均匀缩小,直至达到原来的血管直径,故呈梭形。这种动脉较少发生附壁血栓。
3.圆柱状动脉瘤 开始血管突然呈滚筒状扩张,同样又突然过渡于正常血管。可发生附壁血栓。
4.舟状动脉瘤血管壁呈一侧性扩张,而对侧血管壁则无变化;常见于夹层动脉瘤时。
5.蜿蜒状动脉瘤 相近的血管段相继呈不对称性扩张,因此,被累血管呈蜿蜒状膨隆。大多见于血流方向一再改变的血管(如骨盆的动脉)。
二、根据动脉瘤壁的结构分类
1.真性动脉瘤(aneurysma verum)其壁由所有三层血管壁组织构成,大多数动脉瘤属于此种类型。
2.假性动脉瘤(aneurysma spurium)大多由于血管外伤,血液通过破裂处进入周围组织而形成血肿,继而血肿被机化后其内表面被内皮覆盖。因此,假性动脉瘤乃是一种由内皮覆盖的血肿。
3.多从血管树的血流剪应力量强处及血压变动最明显处(升主动脉、主动脉弓)出发,血流从内膜破裂处钻入病理性疏松的中膜(少数乃来自滋养血管的出血),并顺血流方向将中膜纵行劈开,形成一个假血管腔。这种假血管腔可再次破入真血管腔内,血流如同一个迂回旁道。这种动脉瘤的病因、发病机制颇为复杂,可见于先天性血管畸形、代谢性结缔组织疾病(如Marfan综合征,其主动脉中膜的弹性纤维断裂、缺失,胶原和蛋白多糖增多)、甲状腺功能过低时的血管壁蛋白多糖增多、梅毒性主动脉炎及动脉粥样硬化等。
第六节 风湿病
风湿病(rheumatism)是一种与A组β溶血性链球菌感染有关的的变态反应性疾病。病变主要累及全身结缔组织,呈急性或慢性结缔组织炎症,胶原纤维发生纤维素样变性。本病为结缔组织病(connective tissue disease)即胶原病的一种。心脏、关节和血管最常被累及,以心脏病变最为严重。急性期称为风湿热(rheumatic fever),临床上,除有心脏和关节症状外,常伴有发热、毒血症、皮疹、皮下结节、舞蹈症等症状和体征;血液检查,抗链球菌溶血素O抗体滴度增高,血沉加快等。
本病可发生于任何年龄,但多始发于5~14岁儿童,发病高峰为6~9岁。常反复发作,急性期过后,可造成轻重不等的心瓣膜器质性病变。
风湿病多发生于寒冷地区,热带地区少见。在我国,以东北和华北发病较多,是一种常见病。
一、病因和发病机制
风湿病的病因和发病机制尚未完全明了,但其发生与A组β溶血性链球菌的感染有关。本病多发生于寒冷地区,与链球菌感染盛行地区一致。冬、春季气候寒冷而潮湿,易发生急性扁桃体炎、咽峡炎,从而导致风湿病。抗生素的广泛应用不但能预防和治疗咽峡炎、扁桃体炎,而且明显地减少风湿病的发生和复发。
近来研究证明:A组溶血性链球菌胞壁的成分中,M-蛋白和C-多糖具有特异的抗原性。此外,A组溶血性链球菌能产生一些酶,亦具有抗原性,并能破坏相应的底物,如链球菌溶血素O(能分解血红蛋白)、链激酶能激活血中纤维蛋白溶酶原,使之变为纤维蛋白溶酶(分解纤维蛋白)、链球菌透明质酸酶(分解透明质酸)、链道酶(能分解DNA)及链球菌烟酰胺腺嘌呤二核苷酸酶。此外,还能产生链球菌溶血素S(SLS,为脂蛋白,抗原性微弱)。在链球菌感染时,初次接触抗原后约7~10天,即有抗体形成。在急性风湿性关节炎时,在咽峡炎初次发作后10~15天,患者血清抗链球菌抗体滴度明显升高。至今,临床上仍以检测抗链球菌溶血素O作为诊断指标(滴度1:500单位以上为阳性)。
发病机制有以下学说:①链球菌感染学说:认为本病是链球菌直接感染所致,但从病灶中均未能检测或分离出链球菌。②链球菌毒素学说:认为病变是由链球菌毒素(如SLS、SLO、链球菌蛋白酶、C-多糖等)所引起。③变态反应学说:认为病变是由于机体对链球菌抗原产生超敏反应(主要为Ⅲ型变态反应常伴有血内补体减少,两者均不见于风湿热。而且,几乎不能解释Aschoff小体的发生。④自身免疫学说:目前支持者最多。大多数风湿热患者可证明有对心内膜(或心肌原纤维)、平滑肌(如血管壁)、心内膜等起反应的自身抗体。再者,已证明链球菌与组织成分之间存在交叉反应,即M-蛋白与心肌抗原之间(将M-蛋白注射于动物体内,产生的抗体可与心肌内膜起反应)链球菌多糖与心肌糖蛋白之间,以及链球菌透明质酸与软骨的蛋白多糖复合物之间的交叉反应。在活动性风湿性全心炎患者,免疫荧光检查证明,心肌内有弥漫的免疫球蛋白沉积;心瓣膜(主要在闭锁缘)有IgG沉积,然而是否有自身抗体性质尚无定论。总之,体液因素(Ⅲ型超敏反应、自身免疫)、细胞介导免疫及毒素作用都可能参与发病环节。
二、基本病变
风湿热时,病变可累及全身结缔组织,特别是心脏各层均可被累及;小动脉亦往往被侵犯。病变发展过程大致可分为三期:
1.变质渗出期开始是结缔组织纤维发生粘液样变性,可见胶原纤维肿胀(原纤维间水肿,intermicrofibrillary edema),结缔组织基质内蛋白多糖(主要为氨基葡聚糖)增多。HE染色呈嗜碱性,甲苯胺蓝染色呈异染性,即呈红色。继而肿胀的胶原纤维断裂、崩解成无结构的颗粒状物,与基质中的氨基葡聚糖混合在一起,加上免疫球蛋白、有时还有纤维蛋白沉积,致使病灶的染色性状颇似纤维蛋白,因此称为纤维素样变性(纤维素样坏死)。此外,病灶中还有少量浆液和炎症细胞(淋巴细胞、个别中性粒细胞和单核细胞)浸润。此期持续约1个月。
2.增生期亦称为肉芽肿期(granulomatous phase ),其特点是形成具有疾病特征性的风湿性肉芽肿,即Aschoff小体(Aschoff body),对本病具有诊断意义。
Aschoff 小体体积颇小,一般显微镜下才能看见(图8-19),多发生于心肌间质、心内膜下和皮下结缔组织;心外膜、关节和血管等处少见。在心肌间质内者多位于小血管旁,略带圆形或梭形,其中心部为纤维素样坏死灶,周围有各种细胞成分:①Anitschkow细胞:胞浆丰富,嗜碱性,核大,呈卵圆形、空泡状。染色质集中于核的中央,核的横切面状似枭眼;纵切面上,染色质状如毛虫。②Aschoff巨细胞(Aschoff giant cell):含有1~4个泡状的核,与Anitschkow细胞相似,胞浆嗜碱性。以上两种细胞的来源尚有争论,但现代标记技术证明其为巨噬细胞源性。③小体内还有少量淋巴细胞(主要为T细胞)和个别中性粒细胞。此期经过约2~3个月。
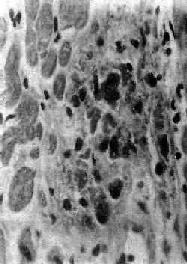
图8-19 Aschoff小体
主由Anitschkow细胞、Aschoff巨细胞和淋巴细胞等组成,中心部常有纤维素样坏死物
3.瘢痕期(愈合期) 细胞成分减少,出现纤维母细胞,产生胶原纤维,并变为纤维细胞。整个小体变为梭形小瘢痕。此期经过约2~3个月。
本病病变的自然经过为4~6个月,但常反复发作,因此,新旧病变常同时并存。
发生在浆膜的风湿病变主要为浆液性和(或)纤维素性炎症。
三、各器官的病变
(一)风湿性心脏病
风湿病时病变常累及心脏各层,故称为风湿性全心炎(rheumatic pancarditis)。
1.风湿性心内膜炎 风湿性心内膜炎(rheumatic endocarditis)常侵犯心瓣膜,其中二尖瓣最常被累及,其次为二尖瓣和主动脉瓣同时受累。三尖瓣和肺动脉瓣一般不被累及。腱索和左心房壁内膜有时也可被侵犯。
病变早期表现为浆液性心内膜炎,瓣膜肿胀、透亮,但尸检时这种早期变化几乎看不到。镜下,瓣膜因浆液性渗出物而变得疏松,伴有巨噬细胞的游入,胶原纤维发生纤维素样坏死。其后,坏死灶周围出现Anitschkow细胞,严重病例可有Aschoff小体形成(图8-20)。几周后,在瓣膜闭锁缘上有单行排列的,直径为1~2mm的疣状赘生物(verrucous vegetation)。此种心内膜炎又称为疣状心内膜炎(verrucous endocarditis)。这些疣赘物呈灰白色半透明,附着牢固,一般不易脱落(图8-21)。镜下,疣赘物为由血小板和纤维素构成的白色血栓。疣赘物主要发生于二尖瓣的心房面和主动脉瓣心室面,其原因:由于瓣膜炎症波及内皮细胞使之受损伤,同时由于心瓣膜不停地关闭和启开,闭锁缘处内膜经常受到摩擦和血流冲击,使受损内皮细胞脱落,内皮下胶原裸露,因而导致血栓形成。有时,左心房壁亦有血栓形成。

图8-20 风湿性心内膜炎
心内膜切片上见增生的细胞呈栅状排列并有Aschoff小体形成
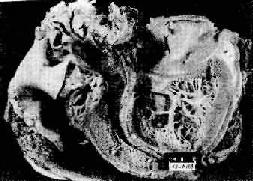
图8-21 风湿性疣状心内膜炎
二尖瓣和主动脉瓣闭锁缘见呈单行排列整齐的细小疣赘物
病变后期,心内膜下病灶发生纤维化,疣赘物亦发生机化。由于风湿病常反复发作,瘢痕形成越来越多。心壁内膜可增厚、粗糙和皱缩,尤以左心房后壁更为显著,称为McCallum斑(McCallum’s patch)。
心瓣膜由于病变反复发作和机化,大量结缔组织增生,致使瓣膜增厚、卷曲、缩短以及钙化,瓣叶之间可发生粘连和纤维性愈着,腱索增粗和缩短,终致形成慢性心瓣膜病(图8-22)。
图8-22 风湿性二尖瓣瓣膜病
瓣膜增厚、变形,瓣叶粘连,腱索增粗、缩短。左心房显著扩张
2.风湿性心肌炎 风湿性心肌炎(rheumatic myocarditis)主要累及心肌间质结缔组织。心肌小动脉近旁的结缔组织发生纤维素样坏死,继而形成Aschoff小体(图8-23)。小体呈弥漫性或局限性分布,大小不一,多呈梭形,最常见于左心室后壁、室间隔、左心房及左心耳等处。后期,小体发生纤维化,形成梭形小瘢痕。
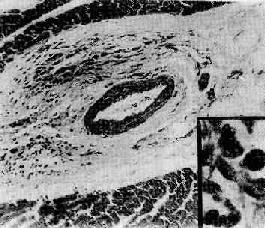
图8-23 风湿性心肌炎
心肌间质中血管旁可见群集的Anitschkow细胞(Aschoff小体),小体已有部分纤维化。右下角为Anitschkow细胞的高倍放大照片,其核状似枭眼
有时在儿童,渗出性病变特别明显,心肌间质发生明显水肿及弥漫性炎性细胞浸润。严重者常引起心功能不全。
风湿性心肌炎常可影响心肌收缩力,临床上表现为心搏加快,第一心间低钝,严重者可导致心功能不全。心电图常见P-R间期延长,可能是由于病变波及房室结或迷走神经兴奋所致。
3.风湿性心包炎 风湿病时,心包几乎总被累及,但临床上,仅15%的风湿性心包炎(rheumatic pericarditis)病例被确诊。病变主要累及心包脏层,呈浆液性或浆液纤维素性炎症,心外膜结缔组织可发生纤维素样变性。心包腔内可有大量浆液渗出(心包积液)。叩诊心界向左、右扩大,听诊时心音遥远,X线检查,心脏呈梨形。当有大量纤维蛋白渗出时,心外膜表面的纤维素因心脏的不停搏动而成绒毛状,称为绒毛心(cor villosum)(图8-24)。恢复期,浆液逐渐被吸收,纤维素亦大部被溶解吸收,少部分发生机化,致使心包的脏、壁两层发生部分粘连,极少数病例可完全愈着,形成缩窄性心包炎(consrictive pericarditis)。
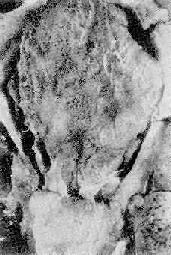
图8-24 风湿性心外膜炎
心外膜上覆盖一层纤维素性假膜,呈绒毛状(绒毛心)
(二)风湿性关节炎
约75%风湿热患者早期出现风湿性关节炎(rheumatic arthritis)。常累及大关节,最常见于膝和踝关节,其次是肩、腕、肘等关节。各关节常先后受累,反复发作,局部出现红、肿、热、痛和功能障碍。镜下,病变主要为浆液性炎,并有少量淋巴细胞和纤维素渗出,有时在关节周围结缔组织内可有少数Aschoff小体形成。愈复时,浆液性渗出物被完全吸收,一般不留后遗症。
(三)风湿性动脉炎
风湿性动脉炎(rheumatic arteritis)可发生于冠状动脉、肾动脉、肠系膜动脉、脑动脉、主动脉和肺动脉等。急性期,血管壁发生粘液样变性和纤维素样坏死,伴有炎症细胞浸润,可有Aschoff小体形成,并可继发血栓形成。后期,血管壁因瘢痕形成而呈不规则增厚,管腔狭窄。
(四)皮肤病变
渗出性病变:躯干和四肢皮肤出现环形红斑(erythema annulare),为环形或半环形淡红色斑,1~2日可消退,发生于风湿热的急性期,对急性风湿病有诊断意义。镜下,红斑处真皮浅层血管充血,血管周围水肿及炎性细胞浸润。
增生性病变:皮下结节(subcutaneous nodules)多见于肘、腕、膝、踝关节附近伸侧面皮下,直径0.5~2cm,圆形或椭圆形,质地较硬,活动,压之不痛。镜下,结节中心为大片纤维素样坏死物质,其周围可见增生的纤维母细胞和Anitschkow细胞呈栅状排列,伴有炎性细胞(主要为淋巴细胞)浸润。数周后,结节逐渐纤维化而变为瘢痕组织。风湿热时,皮下结节并不经常出现,但有诊断意义。
(五)中枢神经系统病变
多见于5~12岁儿童,女孩多于男孩。主要病变为风湿性动脉炎,可有神细胞变性、胶质细胞增生及胶质结节形成。病变主要累及大脑皮质、基底节、丘脑及小脑皮层。当锥体外系统受累较重时,患儿出现肢体的不自主运动,称为小舞蹈症(chorea minor)。
第七节 非感染性心内膜炎
非感染性心内膜炎(non-infective endocarditis)是指不是由病原体直接引起的心内膜炎;在心瓣膜上形成无菌性血栓性疣赘物。风湿性心内膜炎就是一种常见的非感染性心内膜炎。此外,还有以下几种类型。
1.赘疣性血栓性心内膜炎赘疣性血栓性心内膜炎(endocarditis verrucothrombotica)以血液凝固性过高和(或)消耗性血液凝固病为基础,例如可因肿瘤崩解产物(如腺癌的粘液)、休克、内毒素血症和恶病质而引起。因此,相应地又称为肿瘤心内膜炎、休克心内膜炎及消耗性心内膜炎。大多数侵犯左心瓣膜,心房内膜偶尔亦被累及。在心瓣膜闭锁缘形成1mm大或以上的血小板血栓,有时呈息肉状。
2.Libman-Sacks血栓性心内膜炎(Libman-Sacks thrombotic endocarditis) 见于系统性红斑狼疮。心瓣膜(主要见于二尖瓣和三尖瓣)闭锁缘可见一些2~4mm大的粗糙疣状血栓沉积物。
3.Lбffler纤维增生性壁性心内膜炎(endocarditis parietalis fibroplastica Löffler) 颇为少见,几乎只见于男性。特点虽几乎只侵犯心壁内膜,好发于左心室并累及心尖区,但从不侵犯瓣膜和腱索内膜。病因未明,属感染变态反应范畴。早期可见嗜酸性心内膜炎、心肌炎和心包炎,伴有心肌坏死及心壁内动脉炎。坏死物被富含嗜酸性粒细胞的肉芽组织所清除吸收,最后形成瘢痕。被累心内膜可呈白色腱样外观,可并发附壁血栓。心包炎后期可发生纤维化、钙化而形成所谓铠甲心。
第八节 感染性心内膜炎
感染性心内膜炎(infective endocarditis)是指由病原微生物直接侵袭心内膜而引起的炎症性疾病,在心瓣膜表面形成的血栓(疣赘物)中含有病原微生物。引起心内膜感染的因素有:①病原体侵入血流,引起菌血症、败血症或脓毒血症,并侵袭心内膜;②心瓣膜异常,有利于病原微生物的寄居繁殖;③防御机制的抑制,例如肿瘤患者使用细胞毒性药物和器官移植患者用免疫抑制剂时。病因包括各种细菌、真菌及贝纳特考克斯体(coxiella burnettii)等。因此,本病名称宜用感染性心内膜炎,比旧名细菌性心内膜炎更为恰当。临床经过与病原微生物有关,传统分为急性和亚急性两类,其临床经过及病理变化均有所不同。
一、亚急性感染性心内膜炎
亚急性感染性心内膜炎(subacute infective endocarditis)病程经过6周以上,可迁延数月,甚至1~2年。通常由毒力较弱的细菌引起(亚急性细菌性心内膜炎,subacute bacterial endocarditis)。最常见的是草绿色链球菌(streptococcus viridans)(约占75%),此菌为口腔、咽部的正常菌丛。在拔牙、扁桃体摘除术时可有一时性菌血症,细菌亦可从感染灶(牙周炎、扁桃体炎)侵入血流。其次为牛链球菌(为寄居肠道的菌丛)。表皮葡萄球菌为皮肤菌丛,可污染静脉导管及外置起搏器的导线而引起心内膜感染。泌尿生殖器器械检查、前列腺切除术及肠手术后可引起肠球菌性心内膜炎。真菌性心内膜炎最常由白色念珠菌引起,特别是药物成瘾者使用污染的注射器或溶液而造成感染。此外,亦见于免疫抑制的患者。
亚急性感染性心内膜炎常发生于已有病变的瓣膜(如风湿性心内膜炎)或并发于先天性心脏病(如室间隔缺损、Fallot四联症等)。行修补术后的瓣膜亦易被感染。此型心内膜炎最常侵犯二尖瓣和主动脉瓣,并可累及其它部位心内膜。
【病变】
肉眼观,可见在原有病变的瓣膜上形成疣赘物。瓣膜呈不同程度增厚、变形,常发生溃疡,其表面可见大小不一,单个或多个息肉状或菜花样疣赘物。疣赘物为污秽灰黄色,干燥而质脆,颇易脱落而引起栓塞。病变瓣膜僵硬,常发生钙化。瓣膜溃疡较急性感染性心内膜炎者为浅,但亦可遭到严重破坏而发生穿孔(图8-25)。病变亦可累及腱索。
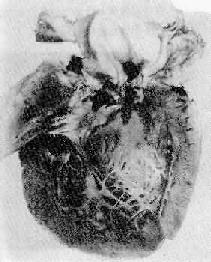
图8-25 亚急性细菌性心内膜炎
主动脉瓣上的赘生物,破坏瓣膜发生穿孔(图中黑色碳棒示穿孔处)
镜检下,疣赘物由血小板、纤维素、细菌菌落、炎症细胞和少量坏死组织构成,细菌菌落常被包裹在血栓内部。瓣膜溃疡底部可见不同程度的肉芽组织增生和淋巴细胞、单核细胞及少量中性粒细胞浸润。有时还可见到原有的风湿性心内膜炎病变。
【结局和合并症】
本病的治愈率较高,但瘢痕形成极易造成严重的瓣膜变形和腱索增粗缩短,导致瓣口狭窄和(或)关闭不全(慢性心瓣膜病)。少数病例可由于瓣膜穿孔或腱索断离而导致致命性急性瓣膜功能不全。
疣赘物内的病原菌可侵入血流,引起败血症,患者皮肤、粘膜和眼底常有出血点,这是由于血管壁受损,通透性升高所致。这种出血在临床诊断上有一定意义。脾一般呈中度肿大,镜下,脾单核吞噬细胞增生,脾窦扩张充血。由于碑功能亢进和草绿色链球菌的轻度溶血作用,患者常有贫血。
动脉性栓塞:瓣膜上的疣赘物颇易脱落,进入血流,可引起各器官的栓塞。动脉性栓塞最多见于脑动脉,其次为肾及脾动脉,冠状动脉栓塞少见。由于栓子多来自血栓的最外层,不含微生物或者由于病原菌毒力弱在局部不能存活,因此多引起非感染性梗死。
免疫性合并症:由于病原菌长期释放抗原入血,可导致免疫复合物形成,伴有补体水平降低。高水平的循环免疫复合物可引起关节炎,指甲下线状出血(subungual splinter haemorrhage)、紫癜及肾小球肾炎。后者大多为局灶性肾小球肾炎,少数病例为弥漫性增生性肾小球肾炎。皮肤可见红色有压痛的小结节(称为Osler结节),但有认为是栓塞性而非免疫性病变。反之,皮肤活检证明,紫癜乃免疫复合物沉积引起的脉管炎所致。脑内小动脉壁可发生纤维素样坏死及炎性细胞浸润,颇似结节性多动脉炎。被累小动脉可发生细菌性动脉瘤(mycotic aneurysm),可并发血栓形成或破裂引起脑出血。
二、急性感染性心内膜炎
急性感染性心内膜炎(acute infective endocarditis)时由于被累心内膜常有溃疡形成,故又称为溃疡性心内膜炎(ulcerative endocarditis)。此类心内膜炎起病急剧,多由毒力较强的化脓菌引起(急性细菌性心内膜炎,acute bacterial endocarditis),其中大多为金黄色葡萄球菌,其次为化脓链球菌。通常病原菌先在机体某局部引起化脓性炎症(如化脓性骨髓炎、痈、产褥热等),当机体抵抗力降低时(如肿瘤、心脏手术、免疫抑制等)病原菌则侵入血流,引起败血症并侵犯心内膜。此型心内膜炎多发生在本来正常的心内膜上,多单独侵犯主动脉瓣,或侵犯二尖瓣。
【病变】
早期,瓣膜闭锁缘上可见污秽黄色脓性渗出物覆盖,瓣膜可被破坏,坏死组织脱落后形成溃疡,其底部多有血栓形成。血栓、坏死组织和大量细菌菌落混合在一起,形成疣赘物(图8-26)。此种疣赘物一般较大,质地松软,灰黄色或浅绿色,易脱落而形成带有细菌的栓子,可引起大循环一些器官的梗死和多发性栓塞性小脓肿(脓毒血症)。严重者,可发生瓣膜破裂或穿孔和(或)腱索断裂,可导致急性心瓣膜关闭不全而猝然致死。镜下,瓣膜溃疡底部组织坏死,有大量中性粒细胞浸润及肉芽组织形成。血栓主由血小板、纤维素构成,混有坏死组织和大量细菌。
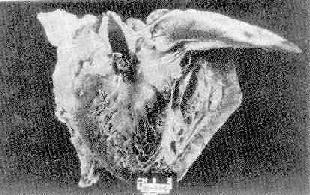
图8-26 急性细菌性心内膜炎
主动脉瓣心室面上的疣赘物(↑)
过去此型心内膜炎病程颇短,患者可在数周至数月死亡。近年来,由于抗生素的广泛应用,死亡率已大大下降。
第九节 心瓣膜病
心瓣膜病(valvular vitium of the heart)是指心瓣膜受到各种致病因素损伤后或先天性发育异常所造成的器质性病变,表现为瓣膜口狭窄和(或)关闭不全。最后常导致心功能不全,引起全身血液循环障碍。心瓣膜病大多为风湿性心内膜炎、感染性心内膜炎的结局。主动脉粥样硬化和梅毒性主动脉炎亦可累及主动脉瓣,引起主动脉瓣膜病,少数是由于瓣膜钙化或先天性发育异常所致。瓣膜关闭不全和瓣膜口狭窄可单独发生,但通常两者合并存在。病变可累及一个瓣膜,或两个以上瓣膜同时或先后受累(联合瓣膜病)。
瓣膜关闭不全(valvular insufficiency)是指心瓣膜关闭时不能完全闭合,使一部分血流返流。瓣膜关闭不全是由于瓣膜增厚、变硬、卷曲、缩短,或由于瓣膜破裂和穿孔,亦可因腱索增粗、缩短和与瓣膜粘连而引起。
瓣膜口狭窄(valvular stenosis)是指瓣膜口在开放时不能充分张开,造成血流通过障碍。主要由于瓣膜炎症修复过程中相邻瓣膜之间(近瓣联合处)互相粘连、瓣膜纤维性增厚、弹性减弱或丧失、瓣膜环硬化和缩窄等引起。
心瓣膜病早期,由于心肌代偿肥大,收缩力增强,可克服瓣膜病带来的血流异常。一般不出现明显血液循环障碍的症状,此期称为代偿期。后来,瓣膜病逐渐加重,最后出现心功能不全,发生全身血液循环障碍,称为失代偿期,此时心脏发生肌原性扩张,心腔扩大,肉柱扁平,心尖变钝,心肌收缩力降低。
一、二尖瓣狭窄
二尖瓣狭窄(mitral stenosis)大多由风湿性心内膜炎所致,少数可由感染性心内膜炎引起。正常成人二尖瓣口开大时,其面积大约5cm2,可通过两个手指。当瓣膜口狭窄时,轻者,瓣膜轻度增厚,形如隔膜。重者,瓣膜极度增厚,瓣口形如鱼口(图8-27)。瓣口面积可缩小到1~2cm2,甚至0.5cm2,或仅能通过医用探针。
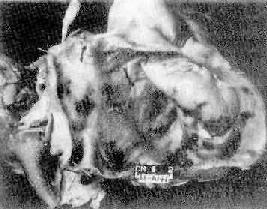
图8-27 慢性风湿性心瓣膜病
二尖瓣口狭窄瓣口呈鱼口状,左心房高度扩张
血流动力学和心脏变化:早期,左心房发生代偿性扩张和肥大。由于二尖瓣口狭窄,舒张期时血液从左心房注入左心室受到障碍,以致舒张末期仍有部分血液滞留于左心房内,加上来自肺静脉的血液,致左心房内血容量比正常增多。此时,心肌纤维拉长以加强收缩力,心腔扩大以容纳更多血液。这种心腔扩大称为代偿性扩张。随着左心房心肌负荷增加,导致其代偿性肥大。后期,左心房代偿失调,心房收缩力减弱而呈高度扩张(肌原性扩张)。此时,左心房血液在舒张期时不能充分排入左心室。由于左心房内血液淤积,肺静脉回流受阻,引起肺淤血、肺水肿或漏出性出血。由于肺静脉血压升高,通过神经反射引起肺内小动脉收缩,使肺动脉血压升高〔正常2kPa(15mmHg),可升高到5.3~6.6kPa(40~50mmHg)。〕
右心的变化:由于长期肺动脉压升高,导致右心室代偿性肥大,心肌纤维增粗。以后,右心室发生心肌劳损,出现肌原性扩张。继而右心房淤血。当右心室高度扩张时,右心室瓣膜环随之扩大,可出现三尖瓣相对性关闭不全。收缩期,一部分血液自右心室回流至右心房,加重右心房的血液淤积,引起大循环淤血。
二尖瓣口狭窄时,左心室内流入血量减少,心室腔一般无明显变化。当狭窄非常严重时,左心室可出现轻度缩小。
临床病理联系:二尖瓣口狭窄,听诊时在心尖区可闻及隆隆样舒张期杂音。X线检查,显示左心房增大。左心房高度扩张时,可引起心房纤维性颤动。左心房血液出现涡流,易于继发附壁血栓,多见于左心房后壁及左心耳内。血栓脱落后可引起栓塞。由于肺淤血、水肿及漏出性出血,肺内气体交换受到影响,患者常咳出带血的泡沫痰,出现呼吸困难、紫绀。患者常出现面颊潮红(二尖瓣面容)。右心衰竭时,大循环淤血,出现颈静脉怒张,各器官淤血水肿,肝淤血肿大,下肢浮肿,浆膜腔积液。
二、二尖瓣关闭不全
二尖瓣关闭不全(mitral insufficiency)也常是风湿性心内膜炎的后果;其次可由亚急性感染性心内膜炎等引起。
二尖瓣关闭不全时,在心收缩期,左心室一部分血液通过关闭不全的二尖瓣口返流到左心房内,加上肺静脉输入的血液,左心房血容量较正常增加,压力升高。久之,左心房代偿性肥大。在心舒张期,大量的血液涌入左心室,使左心室因收缩加强而发生代偿性肥大。以后,左心室和左心房均可发生代偿失调(左心衰竭),从而依次出现肺淤血、肺动脉高压、右心室和右心房代偿性肥大、右心衰竭及大循环淤血。二尖瓣关闭不全与二尖瓣口狭窄相比,除瓣膜的变化不同外,还有左心室代偿性肥大和失代偿后出现的肌原性扩张。X线检查,左心室肥大,听诊时心尖区可闻及吹风样收缩期杂音,其他血液循环变化与二尖瓣口狭窄的相同。后期,瓣膜口狭窄和关闭不全常合并发生。左心房、肺循环、右心及大循环的变化与前述相同,而左心室的增大随着二尖瓣关闭不全的严重程度而愈趋明显。
三、主动脉瓣关闭不全
主动脉瓣关闭不全(aortic insufficiency)主要由风湿性主动脉瓣膜炎造成,也可由感染性主动脉瓣膜炎,以及主动脉粥样硬化和梅毒性主动脉炎等累及主动脉瓣膜引起。此外,梅毒性主动脉炎、类风湿性主动脉炎及Marfan综合征均可引起瓣膜环扩大而造成相对性主动脉瓣关闭不全。
由于瓣膜口关闭不全,在心舒张期,主动脉部分血液返流至左心室,使左心室因血容量比正常增加而逐渐发生代偿性肥大。久之,发生失代偿性肌原性扩张,依次引起肺淤血、肺动脉高压、右心肥大、右心衰竭、大循环淤血。
临床病理联系:主动脉关闭不全,听诊时,在主动脉瓣区可闻舒张期杂音。由于舒张期主动脉部分血液返流,舒张压下降,故脉压差增大。患者可出现水冲脉、血管枪击音及毛细血管搏动现象。由于舒张压降低,冠状动脉供血不足,有时可出现心绞痛。
四、主动脉瓣狭窄
主动脉瓣狭窄(aortic stenosis)主要是慢性风湿性主动脉瓣膜炎的后果,常与风湿性二尖瓣病变合并发生。少数由于先天性发育异常,或动脉粥样硬化引起主动脉瓣钙化所致。
此时,当心收缩期,左心室血液排出受阻,久之,左心室出现代偿性肥大,左心室壁肥厚,但心腔不扩张(向心性肥大)。后期,左心室代偿失调而出现肌原性扩张,左心室血量增加,继之出现左心房淤血。久之,左心房衰竭,引起肺循环、右心功能和大循环障碍。
临床病理联系:X线检查,心脏呈靴形,并向左、向下扩大,向后转位,这是由于其主要病变为左心室肥大的缘故。听诊时,主动脉瓣听诊区可闻吹风样收缩期杂音。严重狭窄者,心输出量极度减少,血压降低,内脏,特别是冠状动脉供血不足。晚期常出现左心衰竭,引起肺瘀血。
第十节 冠状动脉性心脏病
冠状动脉性心脏病(coronary heart disease),简称冠心病,是指因狭窄性冠状动脉疾病而引起的心肌缺氧(供血不足)所造成的缺血性心脏病。冠心病绝大多数由冠状动脉粥样硬化引起。引起冠心病的原因:
1.冠状动脉粥样硬化为最常见的狭窄性冠状动脉疾病,特别是肌壁外冠状动脉支的动脉粥样硬化。冠状动脉近侧段之所以好发动脉粥样硬化是由于它比所有器官动脉都靠近心室,因而承受最大的收缩压撞击。再者,冠状动脉血管树由于心脏的形状而有多数方向改变,因此亦承受较大的血流剪应力。
好发部位:据我国6352例尸检统计,病变的总检出率、狭窄检出率和平均级别均以前降支最高,其余依次为右主干、左主干或左旋支、后降支。性别差异:20~50岁病变检出率,男性显著高于女性;60岁以后男女无明显差异。
病变特点:粥样硬化斑块的分布多在近侧段,且在分支口处较重;早期,斑块分散,呈节段性分布,随着疾病的进展,相邻的斑块可互相融合。在横切面上斑块多呈新月形,管腔呈不同程度的狭窄。有时可并发血栓形成,使管腔完全阻塞(图8-28,图8-29)。根据斑块引起管腔狭窄的程度可将其分为4级:Ⅰ级,管腔狭窄在25%以下;Ⅱ级,狭窄在26%~50%;Ⅲ级,狭窄51%~75%;Ⅳ级,管腔狭窄在76%以上。
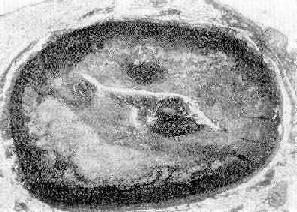
图8-28 冠状动脉粥样斑块并发血栓形成
图示左前降支粥样斑块,累及动脉壁四周,斑块底部粥样物中含大量胆固醇结晶;管腔被血栓阻塞(男性,50岁)
2.冠状动脉痉挛多年来,学术界一直围绕着冠状动脉痉挛是否是缺血性心脏病的原因这一问题进行争论。有人研究心性急死例中发现,其冠状动脉血栓形成的发病率仅为30%,在发作后12小时内死亡的患者中也只占50%,故认为至少有相当部分病例是由于冠状动脉痉挛引起的。近年来,由于心血管造影技术的开展,已证实冠状动脉痉挛可引起心绞痛和心肌梗死。
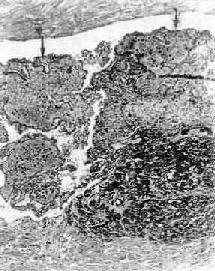
图8-29示图8-28管腔内血栓的放大,主要为白色血栓(↑);右下为纤维素
3.炎症性冠状动脉狭窄冠状动脉的炎症可引起冠状动脉狭窄,甚至完全闭塞而造成缺血性心脏病,例如结节性多动脉炎、巨细胞性动脉炎、高安动脉炎、Wegener肉芽肿病等均可累及冠状动脉。此外,梅毒性主动脉炎亦可造成冠状动脉口狭窄,但都比较少见。
一、心绞痛
心绞痛(angina pectoris)最常见的临床综合征,是由于心肌耗氧量和供氧量暂时失去平衡而引起。心绞痛既可因心肌耗氧量暂时增加超出了已狭窄的冠状动脉供氧能力而发生(劳力型心绞痛)(例如可以体力活动、情绪激动、寒冷、暴饮暴食等的影响下发作),亦可因冠状动脉痉挛导致心肌供氧不足而引起(自发型心绞痛)。
心绞痛的发生是由于心肌缺氧而造成代谢产物的堆积,这些物质刺激心脏局部的交感神经末梢,住处由传入神经经下段颈及上段胸交感神经节和相应脊髓段送至大脑后在相应脊髓段的脊神经所分布的皮肤区域产生不适感,其性质往往不是疼痛而是憋闷或紧缩感。所以,心绞痛是心肌缺血所引起的反射性症状。
1.稳定性心绞痛稳定性心绞痛(stable angina pectoris)亦称为轻型心绞痛,此时心绞痛一般不发作,可稳定数月,仅在重体力劳动时发作。此类心绞痛是由于暂时性急性或慢性相对性心肌缺血所引起。
2.不稳定性心绞痛不稳定性心绞痛(instable angina pectoris)临床上颇不稳定,可在负荷时或休息时发作,或其强度和(或)频度增加。此类患者大多至少有1支冠状动脉大支近侧端高度狭窄。在心绞痛强度增加的病例常见冠状动脉主干和3支冠状动脉狭窄。镜下,常见到因弥散性心肌细胞坏死引起的弥漫性间质性心肌纤维化,可导致慢性肌原性心功能不全,伴有左心室扩张。
3.变异型心绞痛变异型心绞痛(variant angina pectoris)亦称为Prinzmetal心绞痛,多无明显诱因而在休息时发作,仅少数在工作负荷中发病。发作时心电图见ST段反而升高。血管造影证明,此型心绞痛时可见到冠状动脉痉挛,直至其管腔狭窄。这种血管痉挛大多发生在有明显狭窄的冠状动脉,但有时也可见于冠状动脉无明显病变的患者。
二、心肌梗死
心肌梗死(myocardial infarction)是指由于绝对性冠状动脉功能不全(coronary insufficiency),伴有冠状动脉供血区的持续性缺血而导致的较大范围的心肌坏死。绝大多数(95%)的心肌梗死局限于左心室一定范围,并大多累及心壁各层(透壁性梗死),少数病例仅累及心肌的心内膜下层(心内膜下梗死)。
(一)心内膜下心肌梗死
心内膜下心肌梗死(subendocardial myocardial infarction)的特点是坏死主要累及心室壁内层1/3的心肌,并波及肉柱和乳头肌。常表现为多发性小灶状坏死,坏死灶大小为0.5~1.5cm。病灶分布常不限于某1支冠状动脉的供血范围,而是不规则地分布于左心室四周。最严重的病例,坏死灶扩大融合而成为累及整个心内膜下心肌的坏死,称为环状梗死(circumferential infarction)。患者通常存在3大支冠状动脉严重的狭窄性动脉粥样硬化,但绝大多数既无血栓性,亦无粥瘤性阻塞,说明严重、弥漫的冠状动脉病变是此型心肌梗死发生的前提。当患者由于某种原因(如休克、心动过速、不适当的体力活动)引起冠状动脉供血不足时,可造成各支冠状动脉最远端区域(心内膜下心肌)缺氧,而三大支冠状动脉已陷于严重狭窄,侧支循环几乎不能改善心肌的供血,因而导致心肌坏死,而且是多发性小灶状坏死。
(二)区域性心肌梗死
区域性心肌梗死(regional myocardial infarction)亦称为透壁性心肌梗死(transmural myocardial infarction),为典型的心肌梗死类型。梗死区大小不一,多为数厘米大小,或更大些。大多位于左心室,且多累及心壁三层组织。
【病因和发病机制】
1.冠状动脉血栓形成 由于在许多尸检例中发现供养梗死区的冠状动脉支有狭窄性动脉粥样硬化并发闭塞性血栓形成,因此,许多学者认为,冠状动脉血栓形成是心肌梗死的原因。然而,心肌梗死例中冠状动脉血栓的发生率各家报道相差悬殊,因此,关于冠状动脉血栓形成与心肌梗死的关系问题至今仍有争论。
2.冠状动脉痉挛 由于心血管造影技术的进展,使冠状动脉痉挛问题获得突破。现已证实,变异型心绞痛是由冠状动脉痉挛所引起。有人用冠状动脉造影术研究大量的透壁心肌梗死病例,发现冠状动脉闭塞率随发作后时间的延长而递减,提示冠状动脉痉挛的解除。近来研究证明,有严重狭窄的冠状动脉仍可发生收缩。
3.心肌供血不足 在狭窄性冠状动脉粥样硬化的基础上,由于过度负荷而造成心肌供血不足,亦可引起心肌梗死。
【好发部位和范围】
此型心肌梗死的部位与闭塞的冠状动脉供血区域一致。由于左冠状动脉比右冠状动脉病变更为常见,所以心肌梗死多发生在左心室。其中左心室前壁、心尖部及室间隔前2/3,约占全部心肌梗死的50%,该区正是左冠状动脉前降支供血区;约25%的心肌梗死发生在左心室后壁、室间隔后1/3及右心室,此乃右冠状动脉供血区;此外见于左心室侧壁,相当于左冠状动脉回旋支供血区域。
冠状动脉的分布变异较大,据统计有以下3型:①右优势型:右冠状动脉除发出后降支外,还分支供养右心室膈面及左心室一部分。②均衡型:两心室的膈面各由本侧冠状动脉供血,互不越过两心房、两心室交界,并可有两支后降支。③左优势型:左冠状动脉分布于左心室膈面,发出后降支,有的还分支至右心室膈面的一部分。这种变异不但影响心肌梗死的分布,而且,当一支优势的冠状动脉阻塞时,另一支较弱的冠状动脉的吻合支往往不能满足阻塞支供血区心肌对氧的需要,而致该区发生坏死。
【病变】
肉眼观,心肌梗死灶形状不规则。一般于梗死6小时后肉眼才能辨认,梗死灶呈苍白色,8~9小时后呈黄色或土黄色,干燥,较硬,失去正常光泽(图8-30)。第4天在梗死灶周边出现明显充血、出血带。2~3周后由于肉芽组织增生而呈红色。5周后梗死灶逐渐被瘢痕组织取代,呈灰白色(陈旧性梗死灶)。
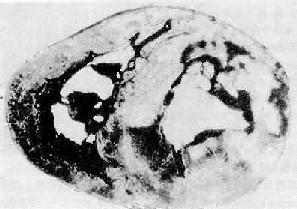
图8-30 心肌梗死
图示心脏横断面由下向上观,图上方为心前壁,左心室前壁和室间隔前部梗死(图中灰白色区)
镜下,心肌梗死最常表现为凝固性坏死,心肌细胞胞浆嗜伊红性增高,继而核消失。肌原纤维结构可保持较长时间,最终融合成均质红染物(图8-31)。梗死灶边缘可见充血带及中性粒细胞浸润,在该处,可见到心肌细胞肿胀,胞浆内出现颗粒状物及不规则横带(图8-32)。另一部分心肌细胞则出现空泡变性,继而肌原纤维及细胞核溶解消失,残留心肌细胞肉膜,仿佛一个空的扩张的肉膜管子(图8-31)。

图8-31 心肌梗死
图左边示心肌纤维肿胀,横纹消失,伊红浓染,核溶解消失(凝固性坏死);右边心肌细胞原纤维及部分心肌核溶解消失(液化性肌溶解)
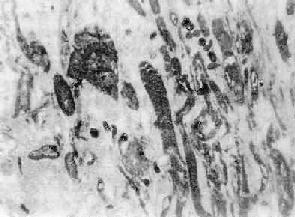
图8-32 心肌梗死
梗死灶边缘的心肌细胞肿胀,胞质内出现颗粒状物及宽窄不等的横带,半薄切片,甲苯胺蓝染色
【心肌梗死的生化变化】
梗死的心肌细胞内糖原减少或消失出现较早,一般在冠状动脉闭塞5分钟后即可出现。这是由于某一支冠状动脉阻塞后,该部分心肌所需的氧和葡萄糖来源中断,细胞内储存的糖原发生酵解所致。近来有报道,心肌缺血早期可引起心肌肌红蛋白缺失。心肌受损时,肌红蛋白迅速从肌细胞释出,进入血液,并从尿中排出,因此急性心肌梗死时能很快从血和尿中测出肌红蛋白值升高。心肌坏死时,一些酶,如谷氨酸-草酰乙酸转氨酶(GOT)、谷氨酸-丙酮酸转氨酶(GPT)、肌酸磷酸激酶(CPK)及乳酸脱氢酶(LDH),可释放入血,使这些酶在血中的浓度升高。其中尤以CPK对心肌梗死的临床诊断颇有帮助。
【合并症及后果】
1.心脏破裂 较少见,约占心肌梗死所致死亡例的3%~13%。常发生在心肌梗死后1~2周内,主要由于梗死灶周围中性粒细胞和单核细胞释出的蛋白水解酶以及坏死的心肌自身溶酶体酶使坏死的心肌溶解所致。好发部位为:①左心室前壁下1/3处,心脏破裂后血液流入心包,引起心包填塞而致急死。②室间隔破裂,左心室血流入右心室,引起右心功能不全。③左心室乳头肌断裂,引起急性二尖瓣关闭不全,导致急性左心衰竭。
2.室壁瘤(ventricular aneurysm)大约10%~38%的心肌梗死病例合并室壁瘤,可发生于心肌梗死急性期,但更常发生在愈合期。由于梗死区坏死组织或瘢痕组织在室内血液压力作用下,局部组织向外膨出而成。多发生于左心室前壁近心尖处,可引起心功能不全或继发附壁血栓。
3.附壁血栓形成(mural thombosis)多见于左心室。由于梗死区心内膜粗糙,室壁瘤处及心室纤维性颤动时出现涡流等原因,为血栓形成提供了条件。血栓可发生机化,或脱落引起大循环动脉栓塞。
4.心外膜炎 心肌梗死波及心外膜时,可出现无菌性纤维素性心外膜炎。
5.心功能不全 梗死的心肌收缩力显著减弱以至丧失,可引起左心、右心或全心充血性心力衰竭,是患者死亡最常见的原因之一。
6.心源性休克 有人认为,当左心室梗死范围达40%时,心室收缩力极度减弱,心输出量显著减少,即可发生心源性休克,导致患者死亡。
7.机化瘢痕形成 心肌梗死后,若病人仍然存活,则梗死灶被机化修复而成瘢痕。小梗死灶约需2周,大梗死灶约4~6周即可机化。
三、冠状动脉性猝死
冠状动脉性猝死(sudden coronary death)较为常见。多见于30~49岁的人,男性比女性多3.9倍。发病有两种情况:①在某种诱因作用下发作:如饮酒、劳累、吸烟、运动、争吵、斗殴等。患者可突然昏倒在地、四肢肌肉抽搐、小便失禁,或突然发生呼吸困难、口吐泡沫、大汗淋漓,很快昏迷。症状发作后迅即死亡,或在1至数小时死亡。②在夜间睡眠中发病:多在家中或集体宿舍中死亡,且往往不被人察觉,所以多无目击者。
引起猝死的原因:多数病例在1支或2支以上冠状动脉有狭窄性动脉粥样硬化,其中有的病例并发血栓形成。据一项尸检报道,在128例冠状动脉性猝死中,89例有较为严重的冠状动脉粥样硬化,18例并发血栓形成,17例有斑块内出血。然而,部分病例冠状动脉仅有轻度,甚至无动脉粥样硬化病变,这部分病例猝死的发生可能是由于冠状动脉痉挛而引起。此外,冠状动脉畸形(如左冠状动脉起源于右冠状动脉窦或一支冠状动脉主干起源于肺动脉干等)、梅毒性主动脉炎所致的冠状动脉口狭窄或闭塞,以及感染性心内膜炎时,主动脉瓣或二尖瓣上的血栓物质脱落,进入冠状动脉口所致的冠状动脉栓塞等均可引起猝死。
第十一节 心肌病
近年来,临床和病理学家将原因不明而又非继发于全身或其他器官系统疾病的心肌原发性损害定名为原发性心肌病(primary cardiomyopathy)。它是非风湿性、非高血压性、非冠状动脉性心肌结构和功能的病理改变。其病理过程属于代谢性而非炎症性,在发病机制上与其它已知病因引起的心脏病无关。相反,若心肌病变与已知病因有关,或继发或伴发于某种全身性疾病时,则称为继发性心肌病(secondary cardiomyopathy)。原发性心肌病较少见,但分布于世界各地。对本病的概念、定义和病理变化等还有不同的认识,分类也较混乱。
心肌是心脏舒缩的动力结构,对物理性(如缺氧)、化学性(如药物、毒素)和生物性(如感染因子)等致病因素特别敏感。心肌的轻度损伤常表现为细胞核及细胞器的肥大,重度损伤表现为细胞结构的改建和细胞坏死,以及由此而导致的纤维化。这些变化既是各型心肌病的基本病变,又具有代偿适应意义。
一、原发性心肌病
1980年WHO将原发性心肌病分为三型:
(一)扩张性心肌病
扩张性(充血性)心肌病〔dilated (congestive)cardiomyopathy〕是原因不明的各种心肌疾病的最后结局,以心腔高度扩张和明显的心搏出力降低(心力衰竭)为特征。大多数病例可查出抗心内膜的自身抗体,其发病学意义尚不清楚。发病年龄为20~50岁,男多于女。患者多因两侧心力衰竭而就医。多数患者常因心力衰竭进行性加重而死亡或因心律心常而发生猝死。
【病变】
肉眼观,典型变化是两侧心室肥大,四个心腔扩张,心尖部变薄呈钝圆形(离心性肥大),状如牛心(图8-33)。重量比正常增加25%~50%(心重400~750g),由于心腔扩张,左心室壁厚度多在正常范围内;右心室壁常轻度增厚。心内膜纤维化在儿童患者较为明显,常伴有心内膜纤维弹性组织增生症。附壁血栓机化后可导致斑块状心内膜纤维化。由于左、右心室扩张,瓣环扩大,可导致二尖瓣及三尖瓣关闭不全。
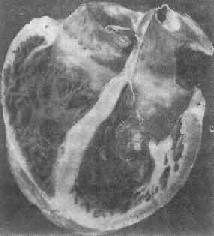
图8-33 扩张性心肌病
两侧心室扩张,壁高度变薄(采自Becker和Anderson)
镜下,心肌细胞通常显示肥大和不同程度的伸长及肌浆变性,失去收缩成分。肥大的心肌细胞由于整个细胞的伸长,其横径多在正常范围,但其核大、浓染。心肌间质纤维化是此型心肌病最常见的变化,可见到血管周围和心肌细胞周围纤细的胶原纤维束,或致密的代替性纤维化灶。间质性纤维化通常以左心室心内膜、心肌为重。心内膜纤维化通常较轻,但附壁血栓处纤维化明显。此外,有些病例可见到淋巴细胞性间质性心肌炎,其特点是多发性淋巴细胞浸润灶伴有心肌细胞的变性和坏死。
(二)肥厚性心肌病
肥厚性心肌病(hypertrophic cardoiomyopathy)的特点是室间隔不匀称肥厚,心肌细胞异常肥大,排列方向紊乱以及收缩期二尖瓣帆向前移动等。肥厚的肌壁顺应性降低,致使心室充盈阻力增加。临床表现为不同程度的心室排空受阻而非充盈受限。根据左心室流出道有无梗阻现象可将其分为梗阻性和非梗阻性两型。右心室流出道或两心室流出道均受阻者少见。常导致猝死,亦可并发感染性心内膜炎。
【病变】
肉眼观,两侧心室显著肥大,心脏重量增加,为正常平均心重的1~2倍(成人患者平均心重582g,少数可达1000g)。绝大多数病例的室间隔厚度大于左室游离壁(图8-34,图8-35),肥厚可为局限性,可累及心基底部(主动脉瓣下)、室间隔中部或心尖区。收缩期二尖瓣向前移动与室间隔接触的结果,可导致二尖瓣增厚和主动脉瓣下心内膜纤维化。在心力衰竭发生之前,左心室一般不扩张。
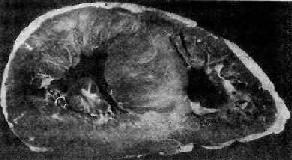
图8-34 肥厚性心肌病
图示心脏横切面(上面观),不对称性心室间隔肥厚(采自Becker和Anderson)
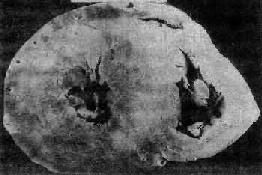
图8-35 肥厚性心肌病
心室横断面(上面观),左心室腔缩小,心室壁均匀增厚,整个心肌布满白色纤维化区,室间隔与后壁厚度之比仅稍大于正常值(1.4)
镜下,心肌细胞显著肥大,核大而浓染(图8-36),核周有亮区包围,组织化学染色证明为糖原堆积,具有一定的诊断意义。心肌细胞排列紊乱较其它型心肌病为甚,而且常呈旋涡状或缠绕呈簇状排列(图8-37),细胞内肌原纤维不呈平行排列,而是向各个方向、互相交错排列。常有间质纤维化灶形成,但以内膜纤维化,尤其位于主动脉瓣下区的内膜纤维化为突出。位于肥厚的室间隔内的冠状动脉分支管壁常有增厚现象。
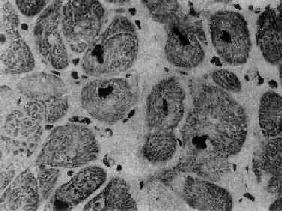
图8-36 肥厚性心肌病
心肌细胞肥大,核大浓染 (采自Edwards)

图8-37 肥厚性心肌病
心肌细胞排列紊乱 ×180(原放大)(采自Edwards)
电子显微镜观察,主要表现为相邻细胞的侧面出现细胞间连接的闰盘;发自某一肌原纤维Z带的肌丝可以不同角度插入另一肌原纤维的Z带,形成交织状排列。有时还可见肌丝从增宽的Z带向各个方向放射。但是,肌细胞侧对侧联接和肌原纤维排列紊乱并非肥厚性心肌病所独有的变化。
(三)限制性心肌病
限制性心肌病(restrictive cardiomyopathy)是以心室充盈受限制为特点。典型病变为心室内膜和内膜下心肌进行性纤维化,导致心室壁顺应性降低,心腔狭窄。因此亦称为心内膜心肌纤维化(endomyocardial fibrosis)。
【病变】
肉眼观,右心室内膜纤维化,尤以心尖部为明显,内膜增厚约2~3mm,灰白色,表面可有血栓形成。心尖部内膜纤维性增厚向上蔓延,可将乳头肌、肉柱埋陷在内,腱索变粗,缩短,可导致三瓣关闭不全。左心室内膜纤维化主要在流入道或心尖部,表面亦可有血栓形成。当二尖瓣后瓣叶与左心室后壁粘连时,引起二尖瓣关闭不全。
镜下,可见增厚的内膜主要为致密的玻璃样变的胶原纤维,可有钙化。表面可见陈旧的附壁血栓。心内膜下心肌常见萎缩、变性改变。
此外,嗜伊红细胞性心内膜心肌病(eosinophilic endomyocardial disease)可能是本型心肌病的亚型。浸润于心内膜心肌处的嗜伊红细胞脱颗粒,释出阳离子蛋白,可引起该处心肌的灶状坏死及纤维化。心室流入道及心尖部的附壁血栓和纤维化可导致心室限制性充盈障碍。
二、克山病
克山病(Keshan disease)是一种地方性心肌病(endemic cardiomyopathy)。1935年首先流行于黑龙江省克山县,当时对该病的本质认识不清,遂以此地名来命名,一直沿用至今。本病主要流行于我国东北、西北、华北及西南一带交通不便的山区或丘陵地带。病理学上以心肌的变性、坏死及修复后形成瘢痕为特点。临床上常有急性或慢性心功能不全表现。
【病因】
50年代以来对本病病因做了大量研究,但至今尚无定论。起初认为是一种地区流行性病毒性心肌炎,可能与柯萨奇B族病毒感染有关,但病毒分离和血清学检测未获规律性阳性结果。近年来发现病区粮食中硒含量明显低于非病区,患者的头发和血液中硒含量亦明显低于非病区人群。服用亚硒酸钠可控制一部分克山病的发作。1974~1977年间,我国在发病地区进行补硒的预防研究,结果表明,经补硒的儿童其克山病的发病例数显著低于对照组。应用硒制剂防治克山病在我国取得了一定的成果,然而,其它可能的致病因素尚有待进一步研究。
【病变】
本病的病变主要在心肌,可出现严重的变性、坏死及瘢痕形成。骨骼肌亦可有轻度变性或小灶状坏死。
肉眼观,心脏呈不同程度增大,两侧心室均扩张,致使心脏近于球形。重量亦增加,病程较长的慢性型病例心重量增加更甚,最重的可超过500g。心腔的扩张属肌原性,心壁变薄,乳头肌及肉柱变扁。少数病例在左心室肉柱间及左、右心耳内可有附壁血栓形成。若血栓脱落,可引起肺、肾、脾、脑等器官的栓塞和梗死。切面上见正常红褐色心肌内散布着数量不等的变性、坏死乃至瘢痕病灶。早期,坏死灶呈灰黄色,境界不清。瘢痕病灶呈灰白色、半透明,境界较清楚,呈星状或树枝状条纹,互相连接,有的呈较大的片块状或带状。心肌病变新旧交杂,色泽斑驳(图8-38)。
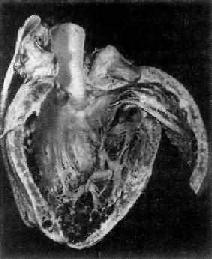
图8-38 克山病之心脏
左心室心肌有一些坏死灶及瘢痕形成,心室内可见附壁血栓
镜下,心肌细胞出现不同程度水肿,表现为胞浆内出现蛋白质颗粒(线粒体肿胀)和空泡变性。心肌坏死主要表现为凝固性坏死和液化性肌溶解。前者肌原纤维融合成均质红染物,核消失,继而坏死物通过细胞自身的及吞噬细胞的溶酶体酶溶解吸收,后者是在心肌空泡变性的基础上,肌原纤维及核发生酶性溶解液化,遗留肉膜空鞘。心肌坏死常呈灶状,病灶大小和形状不一(图8-39),呈散在分布,并多见于心肌内层,而且与冠状血管有密切关系,有的病变围绕冠状动脉分支呈袖套状分布(图8-40)。坏死灶最终被修复而成为瘢痕。
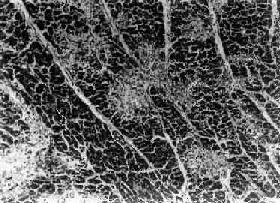
图8-39 克山病之心肌
心肌内可见多数坏死灶
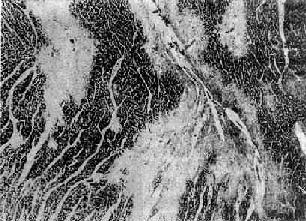
图8-40 克山病之心肌坏死
坏死灶沿血管分布,坏死灶内肌纤维溶解消失
【临床表现】
根据患者发病缓急、病程长短及心肌代偿情况分为4型:
1.急性型 发病急骤,由于心肌病变比较广泛、严重,心肌收缩力明显减弱,心输出量在短时间内大幅度减少,重者出现心源性休克。由于供血不足,患者常有头昏、恶心、呕吐等症状。血压下降,心音弱,尤以第一心音减弱为著,并常有心律不齐。
2.亚急性型病情进展稍缓,心肌受损不如急性型那样严重,但心肌收缩力明显减弱。临床上出现明显的心力衰竭,特别是急性左心衰竭,有咳嗽、呼吸困难、满肺水泡音等征象。约经1~4周后,可发生全心衰竭,出现颈静脉怒张、肝肿大及全身水肿等。
3.慢性型 亦称痨型,病情发展缓慢,多由潜在型逐渐发展而成,少数由急性型或亚急性型转化而来。心脏代偿肥大,心腔扩张明显,临床上主要表现为慢性心功能不全。
4.潜在型 心脏受损较轻或因代偿功能较好,临床上多无明显的自觉症状。
第十二节 心肌炎
心肌炎(myocarditis)是指由各种原因引起的心肌的局限性或弥漫性炎症。虽然某些心肌炎由于在终期可过渡为充血性或限制性心肌病,而被某些学者视为继发性心肌病,但在发病学上心肌炎毕竟是可区分的疾病类型。引起心肌炎的原因很多,诸如病毒、细菌、真菌、寄生虫、免疫反应,以及物理、化学因素等均可引起心肌炎。心肌炎的分类颇不一致,兹根据病因将其常见类型分述如下:
一、病毒性心肌炎
病毒性心肌炎(viral myocarditis)颇为常见,是由亲心肌病毒引起的原发性心肌炎症,常累及心包,引起心包心肌炎。事实上,所谓特发性心肌炎极可能是因病毒感染引起。
【病因和发病机制】
可引起心肌炎的病毒种类颇多,其中最常见的是柯萨奇(Coxsackie)病毒、ECHO病毒(即人肠孤病毒)、风疹病毒、流行性感冒病毒、腮腺炎病毒等。由于在妊娠最初3个月内感染柯萨奇病毒和风疹病毒时可引起胎儿的先天性心脏畸形,因此,这两种病毒占有特别重要的地位。人类的心肌炎以柯萨奇病毒B组感染最为常见。一般而言,亲心肌病毒可直接破坏心肌细胞,但也可通过T细胞介导的免疫反应间接地破坏心肌细胞。由于此类病毒衣壳的糖蛋白分子结构与心肌细胞膜的糖蛋白相似,故在感染后,机体所产生的抗体(激活补体的抗体及中和病毒的抗体)既针对病毒,亦针对心肌细胞。因此,当细胞毒性T细胞被致敏后,即可破坏被病毒感染的心肌细胞。
【病变】
本病病变依患者年龄不同而有所不同。妊娠最初3个月的胎儿感染风疹病毒时,可引起心内膜下心肌的无反应性心肌细胞坏死。在妊娠后期,胎儿感染柯萨奇病毒时则可引起全心炎,大多伴有心骨膜纤维弹性组织增生。初生儿的病毒性心肌炎可见到心肌细胞坏死及粒细胞浸润。其后,代之以巨噬细胞、淋巴细胞、浆细胞浸润及肉芽组织形成(图8-41)。在成人,多累及心房后壁、室间隔及心尖区,有时可累及传导系统。镜下,主要病变为坏死性心肌炎。晚期,可见到明显的心肌间质纤维化,伴有代偿性心肌肥大及心腔扩张(充血性心肌病)。
图8-41 病毒性心肌炎
心肌间质内有弥漫性淋巴细胞及组织细胞浸润
二、细菌性心肌炎
细菌性心肌炎(bacterial myocarditis)可由细菌直接感染,或细菌产生的毒素对心肌的作用,或细菌产物所致的变态反应而引起。
1.心肌脓肿常由化脓菌引起,如葡萄球菌、链球菌、肺炎双球菌、脑膜炎双球菌等。化脓菌来源于脓毒败血症时的转移性细菌菌落,或来自细菌性心内膜炎时的化脓性血栓栓子。肉眼观,心脏表面及切面可见多发性黄色小脓肿,周围有充血带。镜下,脓肿内心肌细胞坏死液化,脓腔内有大量脓细胞及数量不等的细菌集落。脓肿周围心肌有不同程度的变性、坏死,间质内有中性粒细胞及单核细胞浸润。
2.白喉性心肌炎白喉杆菌可产生外毒素,一方面可阻断心肌细胞核蛋白体的蛋白质合成,另一方面可阻断肉碱介导的长链脂肪酸运入线粒体,导致心肌细胞脂肪变性和坏死。镜下,可见灶状心肌变性坏死(图8-42),心肌细胞出现嗜酸性变、肌浆凝聚、脂肪变性及肌浆溶解。病灶内可见淋巴细胞、单核细胞及少数中性粒细胞浸润。病灶多见于右心室壁,愈复后形成细网状小瘢痕。有的病例出现弥漫性心肌坏死,可导致心性猝死。
图8-42 白喉性心肌炎
心肌纤维变性及坏死
3.非特异性心肌炎在上呼吸道链球菌感染(急性咽峡炎、扁桃体炎)及猩红热时,可并发急性非风湿性心肌炎。其发病机制尚未明了,可能是由链球菌毒素引起。病变是间质性心肌炎。镜下,心肌间质结缔组织内及小血管周围有淋巴细胞、单核细胞浸润,心肌细胞有程度不等的变性、坏死。
三、寄生虫性心肌炎
寄生虫性心肌炎(parasitic myocarditis)常见的有以下两种类型:
1.弓形虫性心肌炎此型心肌炎由鼠弓形虫(Toxoplasma gondii)感染而引起。人类主要因食入含有包囊的未煮熟肉类而感染。弓形虫进入人体后,经血流到达单核巨噬细胞系统及各种组织,并在细胞内繁殖。弓形虫侵入心肌细胞后很快繁殖,形成集合体,亦称假包囊。心肌细胞很快破裂,病原体进入周围组织。被破坏的心肌细胞周围有淋巴细胞、单核细胞浸润。愈复后有瘢痕形成。约半数患者因心力衰竭致死。
2.Chagas心肌炎此种心肌炎由原虫枯氏锥虫(trypanosoma cruzi)感染引起,流行于拉丁美洲各国。病情严重,死亡率高。可引起灶状或弥散性心肌坏死,周围有淋巴细胞、单核细胞浸润。心腔扩张,心室壁(主要在心尖区)变薄,常形成室壁瘤,伴有心腔内附壁血栓形成。
四、免疫反应性心肌炎
此类心肌炎见于一些变态反应性疾病,如风湿病、类风湿性关节炎、系统性红斑狼疮、结节性多动脉炎等。其中以风湿性心肌炎最为常见,在心肌间质结缔组织内可见到典型的风湿性肉芽肿(详见本章第六节)。
此外,某些药物可引起变态反应性心肌炎,如磺胺、抗生素(青霉素、四环素、链霉素、金霉素等)、消炎药(保泰松、消炎痛)、抗抑郁药(阿密曲替林)、以及抗癫痫药(苯妥因)等。病变主要累及左心室、室间隔。镜检下,常表现为间质性心肌炎。可引起心肌细胞坏死溶解及淋巴细胞、浆细胞以及引人注目的嗜酸性粒细胞浸润。
五、孤立性心肌炎
孤立性心肌炎(isolated myocarditis)亦称特发性心肌炎(idiopathic myocarditis),至今原因不明。因其首先由Fiedler(1899)所描述,又称Fiedler心肌炎。多见于20~50岁的青、中年人。急性型常导致心脏扩张,可突然发生心力衰竭致死。
【病变】
依组织学变化分为两型:
1.弥漫性间质性心肌炎(diffuse interstitial myocarditis)镜下,心肌间质和小血管周围有多量淋巴细胞、浆细胞和巨噬细胞浸润。有时也可见到嗜酸性粒细胞和少量中性粒细胞。心肌细胞较少发生变性、坏死。
2.特发性巨细胞性心肌炎(idiopathic giant cell myocarditis)病变特点是心肌内有局灶性坏死及肉芽肿形成(图8-43)。病灶中心部可见红染、无结构的坏死物,周围有淋巴细胞、浆细胞、单核细胞和嗜酸性粒细胞浸润,混有许多多核巨细胞。巨细胞的形态、大小各异,可为异物型或Langhans型多核巨细胞(图8-44)。
图8-43 特发性巨细胞性心肌炎
图示左心室心肌的炎症及坏死灶(大小不等的灰白色区域),男性,37岁

图8-44 特发性巨细胞性心肌炎
心肌层内可见多数单核细胞和多核巨细胞
第十三节 心包炎
心包炎(pericarditis)可由病原微生物经血道感染或其毒性代谢产物的作用而引起,心肌坏死亦可波及心外膜引起炎症反应;此外,心包炎亦可因外伤而发生。一般而言,心包炎并非独立性疾病,而大多是一种伴发疾病。
一、急性心包炎
急性心包炎(acute pericarditis)大多为渗出性炎症,常形成心包积液,其性质依引起心包炎的原因而有所不同。在一定程度上,根据渗出物的性质可对其基本疾病作出判断。
1.特发性心包炎特发性心包炎(idiopathic pericarditis)为最常见的心包炎类型,其发病率约占所有心包炎的1/3。其病因尚未明了,可能由病毒、变态反应或毒素引起。临床上,急性发病,有发热、胸骨后疼痛,听诊可闻心包摩擦音,并有典型的心电图改变。
【病变】
此型心包炎是一种纤维素性心包炎,依病变的严重程度可形成浆液纤维素性或纤维素性出血性渗出物。镜下,心外膜充血,可见淋巴细胞、浆细胞浸润。1/3病例可复发,可导致心包愈着,形成缩窄性心包炎。
2.感染性心包炎
(1)病毒性心包炎:多数病毒性心肌炎(尤其柯萨奇病毒引起的心肌炎)除心肌病变外,往往引起心包炎症,其病变与特发性心包炎颇为相似,并常发生钙化,形成钙化性缩窄性心包炎。
(2)结核性心包炎:结核性心包炎(tuberculous pericarditis)多见于青年男性,约占所有心包炎的7%。发病潜隐,低热,血沉加速而无白细胞增多。
结核性心包炎大多经血道感染,或由肺门淋巴结结核蔓延而来,经淋巴道感染者少见。
【病变】
此型心包炎多形成浆液性、出血性心包积液,由于慢性炎症使心包组织疏松,积液有时可达1L以上。有的病例可有多量纤维素渗出,心包表面充血、混浊,擦去纤维素,可见大小不等的结核结节。镜下,心外膜及心包壁层均可检出结核结节。心肌大多早期被累及。积液可全部或部分被吸收,心包两层互相粘连,可完全愈着。自采用抗结核化疗以来,心包钙化已极少见到,而在过去则是典型的变化。
(3)化脓性心包炎:由于对细菌感染性疾病早期用抗生素治疗,化脓性心包炎(purulent pericarditis)的发病率已明显降低,仅约占所有心包炎的0.5%,大多见于败血症或脓毒血症。
此型心包炎多由化脓菌,如链球菌、葡萄球菌及肺炎球菌引起,但有时真菌亦可引起本病。
【病变】
多为纤维素性化脓性炎症,常导致心包积脓,可波及心肌。肉眼可见整个心外膜表面被一层厚的纤维素性脓性渗出物覆盖。
3.胶原病性心包炎
(1)风湿性心包炎:风湿热常侵犯心脏,而心外膜几乎总被累及,发生风湿性心包炎,但临床上仅约15%的病例被确诊。病理变化早期多表现为浆液纤维素性心包炎,晚期心包两层可瘢痕化而互相愈着(详见风湿病)。
(2)狼疮性心包炎:系统性红斑狼疮时,心包最常被累及,将近50%病例发生狼疮性心包炎(lupus pericarditis)。最常表现为纤维性心包炎(fibrous pericarditis),亦可为纤维素性或浆液纤维素性心包炎,后两者特别多见于伴有狼疮性肾炎和尿毒症的患者。此种心包炎可出现或不出现症状。镜下,可见心外膜结缔组织纤维素样坏死,伴有炎性细胞浸润和肉芽组织形成,偶见苏木素小体。此类患者常伴有狼疮性心内膜炎。
4.尿毒症性心包炎:约5%急性和30%慢性肾功能衰竭患者并发尿毒症性心包炎(uremic pericarditis),患者的血液残余氮值常超过85.68mmol/L。
从发病机制分析,本病是排泄性心包炎。当肾功能衰竭时,机体内的“尿毒症毒素”(何种毒素尚未定论,可能是尿素、胍、多胺等)大量蓄积,于是心包、胃粘膜及肺的毛细血管床均参与排毒过程,结果导致这些器官的毛细血管内皮细胞受损。
【病变】
此型心包炎为纤维素性炎症。急性期,肉眼观,可见心包表面有很细的纤维素沉积,继而聚集成绒毛状。镜下,心包组织内可见稀疏的中性粒细胞及淋巴细胞浸润。约5天后,富含毛细血管的肉芽组织从心外膜及心包壁层长入纤维素性渗出物内。尿毒症若得到及时治疗,则心包炎可完全恢复,否则可形成瘢痕,心包两层互相愈着。
二、慢性心包炎
临床上,慢性心包炎(chronic pericarditis)指持续3个月以上的心包炎症,多由急性心包炎转变而来;此型心包炎又可分为两型:
1.慢性非缩窄性心包炎引起慢性非缩窄性心包炎(chronic non-constrictive pericarditis)的原因有:结核病、尿毒症及胶原病;真菌亦可引起。此型心包炎多由急性心包炎演变而来,主要表现为持续性心包积液,较少发生心包愈着。由于炎症及瘢痕形成过程破坏了心包的吸收能力,而且富含蛋白质的渗出液由于其渗透压升高而使积液产生增多。然而,由于积液生成缓慢,心包壁能逐渐适应,不致妨碍心脏活动,因此,临床症状不太明显。
2.慢性缩窄性心包炎慢性缩窄性心包炎(chronic constrictive pericarditis)的特点是由于渗出物机化和瘢痕形成(有时还伴有钙化)而导致心包压力持续性升高,妨碍心脏的舒张期充盈。此型心包炎多见于男性,年龄21~40岁。半数病例原因不明,临床上,大多无急性期表现。在已知原因的病例中,多为结核性心包炎,其它感染性或外伤性心包炎较少见。
【病变】
此型心包炎的病变特点是心包愈着。可分为两个亚型:①心包粘连(concretio pericardii):心包两层互相愈着,心包腔被瘢痕组织所闭塞,但无钙化现象。此型心包炎是抗结核治疗后的典型变化。②钙化性心包炎(pericarditis calculosa):慢性缩窄性心包炎中,约半数病例发生钙化。钙盐沉积好发于冠状沟、室间沟、右心室和靠膈部位。瘢痕组织多发生玻璃样变(糖衣样外观),心肌被压萎缩。钙化的发生一般因心包积血引起,亦可由外伤或病毒性炎症演变而来。
第十四节 先天性心脏病
先天性心脏病(congenital heart disease)是指胚胎时期心和大血管发育异常,又称先天性心脏畸形。先天性心脏病是新生儿和儿童时期(特别是4岁以下的儿童)最常见的心脏病。其病因和发病机制尚未完全明了,一般认为主要由于胚胎早期(妊娠5~8周,亦即胚胎心脏发育的最重要时期)母体内存在某些有害因素(如病毒感染等),影响了心脏的正常发育所致。有些先天性心脏病可能与遗传因素有一定关系。
由于胚胎的心脏发育过程和结构比较复杂,故先天性心血管发育畸形的种类较多。常见类型如表8-1:
表8-1 先天性心血管发育畸形的常见类型
| 类 型 | 占先天性心脏病的百分率 |
| 非紫绀型 | |
| 室间隔缺损 | 25%~30% |
| 动脉导管开放 | 17%~20% |
| 房间隔缺损 | 10%~15% |
| 紫绀型 | |
| 法乐四联症 | 8%~15% |
| 大血管移位 | 8%~10% |
| 其他类型 | |
| 主动脉缩窄 | 5%~7% |
| 肺动脉狭窄 | 5%~7% |
| 主动脉口狭窄 | 4%~5% |
此外,二腔心及三腔心、永存动脉干、双主动脉弓、二尖瓣及三尖瓣发育不全、左冠状动脉起源于肺动脉等均属少见。
一、二腔心及三腔心
二腔心和三腔心是房间隔和(或)室间隔未发育的结果。房、室间隔完全未发育即形成一房一室的二腔心。房间隔或室间隔之一未发育者即成三腔心(一房两室或两房一室)。此类心脏畸形由于动脉血与静脉血完全混合,患儿出生即出现紫绀,不久死亡。
二、房间隔缺损
胚胎发育第5周,自原始总心房的左、右两部分之间长出第一隔膜(第一房间隔),从后上向下生长,使两个心房之间仍然开着的部分(称为第一房间孔)逐渐狭窄,并继续向下生长,与心室间隔的心内膜垫愈合而完全封闭。但在第一房间孔完全封闭之前,第一房间隔上部裂开,形成第二房间孔。
胚胎发育第6周,在第一房间隔的右侧长出第二个隔膜(第二房间隔),向前生长一段,正好把第二房间孔像侧幕一样盖住,但从右向左方向的血流仍可通过。第一和第二房间隔构成的通道即为卵圆孔。出生后肺张开,大量血液从肺静脉进入左心房,产生从左心房向右心房的压差,使第一房间隔上部向第二房间隔靠拢,其后通常与之愈合。约25%的幼儿及儿童解剖学上卵圆孔保持开放,但由于左心房的压高于右心房,因此是封闭着的。
1.第二房间隔缺损为卵圆窝内的一个或多个缺口(亦称为卵圆窝缺损),最大者为整个卵圆窝缺损。其发生是由于第一房间隔上部正常形成第二房间孔的生理性裂缝发生在错误的位置或者太大时,则不能被第二房间隔盖住,结果导致有缺陷的第二房间孔存留。因此,实际上并非第二房间隔缺损,而是第一房间隔中的第二房间孔缺损。
出生后由于肺血流量增多,使左心房压力增高而导致左心房向右心房分流(图8-45)。患者无紫绀。缺损较大者,右心因容量负荷增加而导致右心室肥大和肺动脉高压。严重者可引起继发逆向分流(右心房向左心房分流)而导致紫绀。
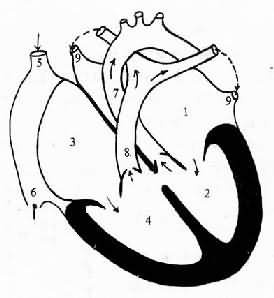
图8-46 室间隔缺损模式图
1.左心房 2.左心室 3.右心房 4.右心室 5.上腔静脉 6.下腔静脉 7.主动脉 8.肺动脉 9.肺静脉
单独室间隔膜部缺损一般不大。在心室收缩期,左心室内压力高于右心室,部分血液分流到右心室内,右心室血液容量因而增加,输入肺循环的血液量也随之增多。这样,由肺静脉回流到左心的血量亦增加,最后可依次导致右心室、肺动脉、左心室、左心房的扩张和肥大。当缺损甚小时,向右心室分流的血液量虽然很少,但是血流通过狭窄的小孔却能发生较大的涡流,临床听诊可闻及明显的收缩期杂音。
四、Fallot四联症
Fallot四联症(tetralogy of Fallot )由Fallot(1888)首先描述。此种心脏畸形有4个特点:①肺动脉流出道狭窄;②室间隔膜部巨大缺损;③主动脉右移,骑跨于室间隔缺损上方;④右心室高度肥大及扩张(图8-47)。
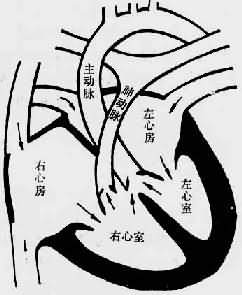
图8-47 Fallot四联症模式图
肺动脉狭窄,室间隔膜样部巨大缺损,主动脉右移并骑跨在室间隔上方,右心室高度肥大及心腔扩张
这种畸形的发生是由于肺动脉肌性圆锥发育障碍伴有狭窄,室上嵴错位和圆锥肌与肌性室间隔不能融合,结果导致高位室间隔缺损,伴有膜部缺损。
肺动脉狭窄多见于肺动脉瓣口,较少见于肺动脉干和肺动脉圆锥部。右心室因血液输入肺受阻而发生代偿性肥大。
室间隔有巨大缺损,心收缩期,部分血液由左心室分流入右心室,以致右心室的血液容量增加,发生代偿性扩张和肥大。
此外,由于主动脉骑跨在室间隔缺损的上方,同时接受左、右心室的大量血液,结果发生管腔扩张和管壁增厚,肺动脉愈狭窄,右心室注入主动脉的血液量亦愈多,主动脉的扩张和肥厚也愈明显。增大的主动脉与狭窄的肺动脉形成鲜明的对比。少数患儿可合并其他心脏畸形,如主动脉位于右侧等。
临床上,患儿有明显紫绀,肺动脉狭窄的程度愈重,紫绀愈明显。这是因为肺动脉高度狭窄时,一方面促使右心室的静脉血更多地分流进入主动脉,另一方面是右心室的血液难以注入肺循环进行气体交换之故。X线检查,右心室高度肥大,肺由于血液输入量减少而显示肺纹理减少,肺野异常透明、清晰。
患儿一般能存活多年,少数可到成年是由于侧支循环的代偿作用。支气管动脉常出现代偿性扩张,肺动脉与支气管动脉之间的侧支循环使主动脉的血液可通过侧支进入肺而得到代偿。极少数病例合并动脉导管开放,扩张的动脉导管成为重要的侧支循环。本病可行手术治疗。
五、动脉导管开放
动脉导管是胎儿期连接肺动脉和主动脉的一条短的动脉管道,在出生时,导管的直径约为主动脉直径的一半,以后逐渐闭锁。生理性闭锁时间一般在出生时,或出生后半年左右,少数可迁延到一年后。
动脉导管开放(patent ductus arteriosus)是指导管完全未闭或仅一部分未闭(图8-48)。此种畸形可单独存在或与其他心脏畸形(房间隔缺损、室间隔缺损、肺动脉狭窄等)合并发生。单纯动脉导管开放时,由主动脉分流到肺动脉的血液量甚多。因为血液是从主动脉(动脉血)流入肺动脉,故患儿无紫绀。单纯动脉导管开放手术结扎可治愈。
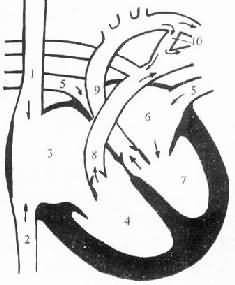
图8-48 动脉导管未闭模式图
1.上腔静脉 2.下腔静脉 3.右心房 4.右心室 5.肺静脉6.左心房7.左心室 8.肺动脉 9.主动脉 10.动脉导管
六、主动脉缩窄
主动脉缩窄(coarctation of the aorta)为非紫绀型先天性心脏病中较常见的一种类型。本病分为幼年型及成人型:
1.幼年型为动脉导管前的主动脉峡部狭窄。狭窄常较重,主动脉血液通过量减少。本型常合并动脉导管开放畸形,肺动脉内一部分静脉血液可经过开放的动脉导管注入降主动脉,因此,患者下肢动脉血液含氧量低,因而严重青紫,而上肢动脉血的含氧量则正常。婴儿出生后,若动脉导管发生闭锁,则不能存活。
2.成人型为动脉导管后的主动脉峡部狭窄。狭窄程度一般较轻,一般动脉导管已闭锁。由于狭窄位于动脉导管闭合口的远侧,所以,胸主动脉与腹主动脉之间存在较高的压差。日久即出现代偿适应现象,表现为主动脉弓部的动脉分支(胸廓的动脉、乳房内动脉及其肋间支)均逐渐扩张并与降主动脉的分支(肋间动脉、腹壁深动脉等)之间发生侧支循环以保证下肢的血液供应。
七、大血管移位
大血管移位(transposition of the great vessels)是主动脉和肺动脉在胚胎发育转位过程中出现的异常,可分为纠正型和非纠正型两种。
1.纠正型主动脉移位于前方,肺动脉移向后侧,两者前后平行排列。然而,通常伴有左右心室的相互移位。因此,主动脉仍出自左心室,肺动脉出自右心室。血液循环正常,患者无症状,可健康存活。
2.非纠正型主动脉与肺动脉互相交换位置,即主动脉出自右心室,而肺动脉出自左心室,主动脉位于肺动脉之右前侧,两者无正常形式的交叉,呈平行排列。右心室的血液不能注入肺进行气体交换,而由主动脉注入大循环中;左心室的血液则不能注入全身,而经肺动脉注入肺。非纠正型(又称完全型)大血管移位,在胚胎期,因有脐静脉,并有动脉导管的沟通,对胚胎发育无大影响。出生后,肺开始呼吸,患儿出现紫绀,若心脏无其他血液通路,出生后很快死亡。出生后尚能存活者,均有其他畸形合并存在,在大、小循环之间出现异常通路,如卵圆孔未闭、动脉导管开放、房间隔缺损和室间隔缺损等。这些异常通路可使部分血液发生混合,供给全身需要,维持生命。
第十五节 心脏肿瘤
心脏肿瘤颇为少见,其中原发性肿瘤更为罕见,转移性肿瘤约为原发性的20~40倍。原发性心脏肿瘤大多为良性,其中又以心房粘液瘤居多数。
一、心脏良性肿瘤
1.粘液瘤心脏粘液瘤约占良性原发性心脏肿瘤的50%。年龄30~50岁,性别无明显差异。临床上,常有血流受阻和栓塞症状。本瘤多发生于左心房(约占75%),发生于右心房的占20%,心室5%。肿瘤大小不等,呈息肉状或绒毛状。切面呈灰白色半透明胶冻状,质软易碎。镜下,粘液瘤细胞呈星芒状或梭形,核呈卵圆形或梭形。瘤细胞稀少,散在或三五成群,分布于大量粘液样基质中,基质内富含蛋白多糖。关于本瘤的组织发生,观点仍不一致,许多学者认为本瘤来源于心内膜下的多能性原始间叶细胞。
2.横纹肌瘤心脏横纹肌瘤多见于15岁以下儿童,约50%病例伴有结节性硬化。临床上,肿瘤小者可无症状,大者可向心腔突起,引起阻塞症状,多发性肿瘤常引起严重的充血性心力衰竭。肉眼观,肿瘤多位于左和右心室的心肌内,常为多发性,直径数毫米至数厘米。镜下,瘤组织疏松,细胞较大(直径可达80μm),呈卵圆形。胞浆空泡状,富含糖原,核居中,核仁明显。核周围的胞浆呈疏网状,细胞形似蜘蛛,故有蜘蛛细胞之称。目前认为本瘤是一种源自胚胎心肌母细胞的婴儿错构瘤。
3.纤维瘤心脏纤维瘤多见于婴儿和儿童。临床上,可引起左、右心室流出道阻塞症状及充血性心力衰竭。肉眼观,肿瘤多位于左心室或室间隔内。多为单发,大小不一,直径有时可达10cm。镜下,与其他部位的纤维瘤相似。
二、心脏恶性肿瘤
心脏原发性恶性肿瘤甚少见,可有恶性血管内皮瘤、横纹肌肉瘤、恶性间皮瘤、纤维肉瘤等。
三、心脏转移性肿瘤
其他部位恶性肿瘤转移至心脏者少见。但亦有人认为,如果仔细检查也并非很少。心脏转移瘤中以恶性黑色素瘤较多。心脏转移瘤可从邻近器官的恶性肿瘤蔓延而来,如支气管癌、胃癌、食管癌和纵隔肿瘤等,但大多系经血道转移而来。
第九章 呼吸系统疾病
呼吸系统包括鼻、咽、喉、气管、支气管和肺,是通气和换气的器官。通常以喉环状软骨为界将呼吸道分为上、下两部分。下呼吸道从气管起,支气管经逐级分支到达肺泡。终末细支气管以上为传导部分,呼吸细支气管以下为换气部分。临床上,通常把管径<2mm的小支气管和细支气管,称为小气道。传导性气道管壁覆盖纤毛柱状上皮,肺泡则由肺泡上皮覆盖。Ⅰ型肺泡细胞胞浆扁阔,覆盖90%以上的肺泡面积。Ⅰ型肺泡上皮细胞和毛细血管内皮细胞及其基膜组成的肺泡毛细血管膜(alveolocapillary membrane)构成血气屏障,是气血交换的场所。Ⅱ型肺泡细胞为数较少,呈立方形,镶嵌在Ⅰ型上皮细胞之间,能排泌肺表面活性物质。
呼吸系统与外界相通,肺又是体内唯一接受全部心输出血量的器官,血流量也多,环境中的有害气体、粉尘、病原微生物及某些致敏原和血流中的致病因子易侵入肺内引起疾病。在以往,呼吸系统疾病中以感染性疾病居多,尤其是细菌性肺炎、肺结核病较常见。随着抗生素的普遍应用,感染性疾病得以被有效控制。而由于大气污染、吸烟和某些其它因素,慢性阻塞性肺疾病、肺癌、职业性肺疾病、慢性肺源性心脏病等的发病率和病死率则日趋增多,应引起足够重视。
尽管呼吸系统与外界环境接触最频繁,环境中的有害因素常是诱发肺疾病的主要原因,但呼吸系统的防御功能,能净化自身,可防止有害因子入侵造成损伤。纤毛-粘液排送系统乃呼吸道特有的保护装置,能将沉积于粘液中的有害因子自下而上地向外排送;而且粘液成分中还含有溶菌酶、干扰素、补体系统、分泌型IgA等免疫活性物质,具有增加局部免疫力的作用。肺泡毛细血管膜不仅能清除沉积于肺内的微粒,还具有选择性渗透的屏障作用,能防止吸入性有害物质侵入肺深部组织。肺巨噬细胞是肺内重要的防御细胞,能吞噬吸入的有害物质,还可摄取和处理抗原,将抗原信息传递给淋巴细胞,以增强其免疫活性,参与特异性免疫反应。呼吸系统防御装置如受损,则防御功能降低,在发病环节中起着重要作用。
第一节 鼻及鼻窦疾病
一、鼻衄
鼻出血亦称鼻衄(epistaxis)颇为常见,一般可分为两类,由鼻局部原因所致的鼻衄和作为全身性疾病的局部表现之症状性鼻衄。尤以症状性鼻衄更为常见,其发病率约为前者的2倍。
局部因素引起的鼻衄大多为单侧性,出血多发生在鼻中隔前部富于毛细血管前动脉的部位(Kisselbach区)。此处组织弹性较低、遭受刺激时(如挖鼻、干燥、粉尘等),粘膜血管容易破裂,导致出血。鼻粘膜溃疡、鼻息肉和鼻腔恶性肿瘤更是引起出血的原因。
症状性鼻衄可见于血管和肾疾病(如血管壁损害、尿毒症等)。出血素质和某些传染病(如流感、麻疹、白喉、伤寒等)。遗传性出血性末梢血管扩张症(Osler病)患者的血管壁先天性薄弱,稍有扩张则可破裂出血。
二、鼻炎
鼻炎(rhinitis)是常见病,有急性和慢性鼻炎两类。
(一)急性鼻炎
根据病因可分为急性病毒性鼻炎和过敏性鼻炎。
1.急性病毒性鼻炎 可由各种呼吸道病毒引起,而以鼻病毒和冠状病毒为主要致病病毒。受凉、过劳、全身慢性病、鼻中隔偏曲等诱因可导致机体抵抗力降低或鼻粘膜防御功能破坏,致病病毒得以入侵并生长繁殖,经1~3天潜伏期而发病。初期,鼻粘膜呈浆液性卡他,继而,由于原来存在于鼻粘膜的链球菌、葡萄球菌等致病菌乘机活跃增殖,形成继发感染,病变转为脓性卡他。2~3天后上皮开始再生,约2周后重新修复而痊愈。但在免疫力和低抗力尚未发育完善的婴幼儿,有时可伴发鼻窦炎、中耳炎、支气管炎、肺炎或急性心肌炎等招致严重后果。
2.过敏性鼻炎 属于Ⅰ型变态反应性疾病。变应原主要是花粉、室内尘埃(特别是尘螨)、动物毛屑等,此外,碘、酒精、油漆、某些食物、药品或化妆品也可引起。镜下可见鼻粘膜上皮层内杯状细胞增多,基底膜增厚,间质水肿,有大量嗜酸性粒细胞、淋巴细胞和浆细胞浸润,肥大细胞也较多。
(二)慢性鼻炎
1.慢性单纯性鼻炎 是基于鼻腔血管的神经调节功能紊乱,导致以鼻粘膜血管扩张、腺体分泌增多为特征的慢性炎症。病变表现为粘膜肿胀,血管扩张充血,粘液分泌增多,血管和腺体周围可见淋巴细胞和浆细胞浸润。
2.慢性肥厚性鼻炎 是以粘膜、粘膜下层以至骨质增生肥厚为特征的慢性炎症。镜下除见粘膜充血水肿、扩张的血管周围有一些圆形细胞浸润外,粘膜下结缔组织增生,使粘膜变得肥厚,甚至形成息肉。深层的鼻甲骨和骨膜亦可增生、肥大。
3.慢性萎缩性鼻炎 是以鼻粘膜萎缩、嗅觉障碍或消失、鼻腔内有痂样苔被覆为特征的慢性炎症。鼻痂常被粘染的腐败菌所分解,产生恶臭,故又称臭鼻症。镜下常见粘膜上皮呈广泛的鳞状化生,血管呈闭塞性脉管炎变化,由于血管闭塞,血液循环不良,导致粘膜、腺体以至骨质萎缩,纤维结缔组织增生(纤维化)。
此外,有些疾病如结核、梅毒、麻风、结节病等可在鼻粘膜形成慢性炎性肉芽肿,
■[此处缺少一些内容]■
、眼窝蜂窝织炎、软脑膜炎、脑脓肿等。严重的化脓性鼻窦炎还可导致败血症。
四、鼻及鼻窦肿瘤
(一)良性肿瘤
鼻腔和鼻窦的良性肿瘤以乳头瘤、纤维血管瘤、血管瘤和骨瘤较为多见,鼻混合瘤、软骨瘤、神经纤维瘤、牙釉质细胞瘤较少。此外亦可发生淋巴管瘤、粘液瘤、腺瘤、平滑肌瘤和脂肪瘤等,但颇为罕见。
(二)恶性肿瘤
鼻部和鼻窦的恶性肿瘤以癌的发病率最高,约为肉瘤的4~9倍。其中以鳞状细胞癌最为常见,基底细胞癌、粘液表皮样癌、腺癌及未分化癌较少。其他恶性肿瘤可有恶性淋巴瘤、髓外浆细胞瘤、纤维肉瘤、恶性黑色素瘤和嗅神经母细胞瘤等。
恶性肉芽肿(malignant granuloma)又称中线恶性网状细胞增生症(midline malignant reticulosis,MMR)或致死性中线肉芽肿(lethal midline granuloma)是鼻部较重要的一种疾病,可见于各级年龄,但在40岁前后发病率最高,且多为男性。患者多有不规则发热、鼻塞、鼻衄及脓涕等症状,鼻部肿胀,鼻内可见坏死、溃疡。病变进一步扩展,可破坏软组织,软骨和骨,导致鼻中隔穿孔和硬腭溃疡,有些患者面部皮肤也可遭破坏。终期患者可出现恶病质。病程短者仅1个月,长者可达10余年,但多数在1年以内。病理组织学上,最突出的病变特征是在病变组织中有大量异型淋巴样细胞并混有数量不等的中性粒细胞、淋巴细胞、浆细胞和单核细胞浸润。同时,有不同程度的凝固性坏死。异型淋巴样细胞大小不等,形态不一,核形不规则,深染或染色质呈细网状,可有一个或多个小核仁,核分裂像较多。这些细胞常可浸润粘膜上皮和血管,受累血管管腔变窄,有的血管腔内还可见血栓形成。用免疫组织化学染色方法,可证明异型淋巴样细胞能表达T细胞分化抗原。目前认为,本病是一种中线粘膜相关淋巴组织来源的周围T细胞性淋巴瘤。近年来的研究发现,在一些T细胞肿瘤的病变组织中或其瘤细胞上存在着EBV-DNA。最近有人用针对EB病毒编码的小分子RNA的寡核苷酸探针(EBER寡核苷酸探针)对MMR病变组织进行原位杂交检测,发现患者EB病毒隐匿感染的检出率为78.9%,故推测本病的发生可能与EB病毒感染有关。
第二节 咽部疾病
一、咽炎
咽炎(pharyngitis)是咽粘膜及其淋巴组织的炎症。急性咽炎常为上呼吸道感染的一部分,多由病毒感染引起。主要致病病毒是柯萨奇病毒、腺病毒和副流感病毒。由细菌感染引起者,致病菌多为链球菌、葡萄球菌和肺炎球菌。病变可表现为急性单纯性咽炎和急性化脓性咽炎。急性脓毒性咽炎(acute septic pharyngitis)由溶血性链球菌引起,局部症状和全身症状以及病变都较严重,甚至发生脓毒败血症。急性咽炎反复发作可转为慢性,长期烟酒过度或受有害气体刺激也可引起慢性咽炎。慢性咽炎可分为:①慢性单纯性咽炎:病变特点是粘膜充血,粘液腺肥大、分泌增多,粘膜层内可见一些圆形细胞浸润,淋巴组织和纤维结缔组织可轻度增生,②慢性肥厚性咽炎:粘膜增厚,淋巴组织和纤维结缔组织明显增生,以致在咽后壁形成颗粒状隆起,③慢性萎缩性咽炎:多由萎缩性鼻炎蔓延而来,此时,粘膜和腺体均萎缩。
二、扁桃体炎
(一)急性扁桃体炎
急性扁桃体炎(acute tonsillitis)常发生于儿童及青年。病原菌主要是乙型溶血性链球菌,此外,葡萄球菌、肺炎链球菌和腺病毒也可引起。按其病变特点可分为:
1.急性卡他性扁桃体炎 病变轻,且多局限于粘膜层,可见炎性细胞浸润,扁桃体充血、肿胀,但肿大不明显,表面也无渗出物。
2.急性化脓性扁桃体炎 扁桃体明显肿大,普遍充血及大量中性粒细胞浸润,隐窝内充满脓性渗出物。病变较重者,多数淋巴滤泡增大、化脓,形成多发性滤泡脓肿,并可向隐窝或表面穿破,形成溃疡,小脓肿也可融合,致使整个扁桃体化脓。
(二)慢性扁桃体炎
慢性扁桃体炎(chronic tonsillitis)多由急性扁桃体炎反复发作演变而来。按其病变可分为:
1.慢性肥大性扁桃体炎 扁桃体呈不同程度肿大,淋巴组织显著增生,淋巴滤泡增多、增大,生发中心扩大,淋巴组织内,特别粘膜上皮下可见大量浆细胞和一些中性粒细胞浸润,隐窝内可见炎性渗出物。有时,由于淋巴组织和结缔组织增生可形成息肉样肿物。
2.慢性纤维性扁桃体炎 由于大量纤维结缔组织增生(瘢痕化),扁桃体体积缩小、质硬,淋巴组织往往萎缩甚至消失。隐窝常因出口阻塞而呈囊状扩张,囊内也可见炎性渗出物。在增生的结缔组织中有时可见软骨或骨化生。
三、咽部肿瘤
(一)良性肿瘤
咽部较常见的良性肿瘤有乳头状瘤、血管瘤、鼻咽纤维瘤、多形性腺瘤等。此外,亦可有神经鞘瘤、神经纤维瘤、淋巴管瘤、脂肪瘤、软骨瘤等,但均较少见。
(二)恶性肿瘤
咽部的恶性肿瘤中鼻咽癌最为常见,而扁桃体癌、恶性淋巴瘤、横纹肌肉瘤较少,平滑肌肉瘤、恶性黑色素瘤均属罕见。
鼻咽癌(nasopharyngeal carcinoma) 是鼻咽部上皮组织发生的恶性肿瘤。在我国,鼻咽癌居各种恶性肿瘤的第8位,在头颈部的恶性肿瘤中鼻咽癌的发病率则居首位,是我国重点防治的十大肿瘤之一。广东、广西、福建、湖南、四川、台湾及香港等地鼻咽癌的发病率较高,男性患者为女性患者的2~3倍,发病年龄多在40~50岁之间。临床上,患者常有涕中带血、鼻塞、鼻衄、耳鸣、听力减退、头痛、颈淋巴结肿大及脑神经受损等症状。
【病因】
鼻咽癌的病因尚未完全查明。可能与环境、遗传、病毒等方面因素有关。经研究发现,有些化学物质,如多环芳烃类、亚硝胺类、微量元素镍等与鼻咽癌有一定关系。动物实验还发现维生素A缺乏,性激素失调等可能提高鼻咽粘膜对化学致癌物质的敏感性。又据调查,有些鼻咽癌患者有家族聚集性和种族易感性。如有些患者的兄弟、姊妹或父母也患或曾患鼻咽癌,还有的孪生兄弟相继发病,提示鼻咽癌可能与遗传因素有关。近年来,有大量研究表明,EB病毒与鼻咽癌有非常密切的关系。90%左右的未分化鼻咽癌组织中可检出EB病毒。癌细胞内存在EBV-DNA和核抗原(EBVA),患者血清中也可出现与EB病毒核抗原、早期抗原、膜抗原和壳抗原相应的抗体。尤其是抗EB病毒壳抗原的IgA抗体(VCA-IgA)阳性率可高达97%,颇为特异而有诊断价值,可作为鼻咽癌早期诊断的参考。EB病毒的存在与鼻咽癌的组织学类型和分化程度密切相关,在泡状核细胞癌和低分化鳞状细胞癌组织中EB病毒的检出率极高,一部分鼻咽低分化腺癌也可发现EB病毒。而且,VCA-IgA抗体水平随病人存活时间延长而逐渐下降,当肿瘤复发或发生远处转移时又复上升,因而对患者治疗后的随访监视和预后估计具有较大的参考价值。
【病理变化】
鼻咽癌最多见于鼻咽顶部,其次是外侧壁和咽隐窝,发生于前壁者最少。但原发癌灶在两个部位(如顶部和侧壁)同时发生的也不少见。
肉眼观:早期病例常表现为局部粘膜粗糙或稍隆起。有些原发癌可形成隆起于粘膜的小结节(图9-1),肿瘤逐渐发展可表现为结节型、菜花型、粘膜下型、浸润型或溃疡型(图9-2)肿块。据统计,结节型占41.4%,菜花型占17.5%,粘膜下型占15.1%,浸润型占12.7%,溃疡型占2%,分型不明者占11.3%。
组织学类型:鼻咽癌多来自鼻咽粘膜柱状上皮,主要是隐窝柱状上皮,少数发生自鼻咽粘膜鳞状上皮,由鼻咽部腺上皮发生者极少。鼻咽癌组织结构复杂,以致分类意见不统一。一般按其分化程度分为高分化、低分化和未分化癌三大类。并按其组织学结构分为:①鳞状细胞癌,②腺癌,③泡状核细胞癌和④未分化癌等四种基本类型。
图9-1 鼻咽癌
箭头示咽扁桃体右侧小瘤结
图9-2 鼻咽癌
鼻咽部正中矢状切面,整个鼻咽部被癌瘤所侵占,并形成巨大溃疡
(1)鳞状细胞癌:高分化鳞状细胞癌的癌巢细胞分层明显,可见清晰的棘细胞及细胞内角化,有的还可见角化珠(图9-3)。低分化鳞状细胞癌的癌巢细胞分层多不明显,癌细胞呈多角形、卵圆形、梭形或不规则形,胞浆丰富,境界清楚,少数细胞可见细胞间桥,但无角化现象。
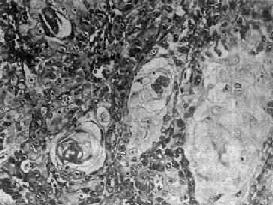
图9-3 鼻咽高分化鳞状细胞癌
癌细胞分化成熟,有角化珠形成
(2)腺癌:高分化腺癌包括柱状细胞腺癌(图9-4)和腺泡状腺癌。低分化腺癌癌细胞呈不规则条索状或片状排列,有时可见腺腔结构或围成腺腔的倾向(图9-5)。有的癌细胞胞浆内含有泡沫状分泌空泡。
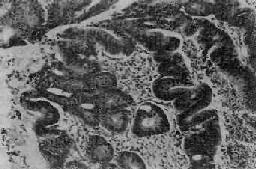
图9-4 鼻咽柱状细胞腺癌
癌瘤从表面柱状上皮(左)发生,向深处浸润生长,并呈腺管状结构
图9-5 鼻咽低分化腺癌
癌巢不规则,部分出现腺腔结构(箭头),癌细胞稍小,核卵圆或圆形,可见核仁
(3)泡状核细胞癌:癌巢不规则,境界不甚明显,癌细胞胞浆丰富,境界不清晰,往往呈合体状聚集成堆。核大呈空泡状,圆形或卵圆形,有1~2个肥大的核仁,核分裂像并不多见(图9-6)。癌细胞间常可见淋巴细胞浸润。电镜下,可观察到紧挨淋巴细胞的癌细胞胞膜和胞质有破坏现象,这可能是淋巴细胞对癌细胞免疫攻击的结果。

图9-6 鼻咽泡状核细胞癌
癌细胞境界不清,呈合体状,核大圆形,染色质少,呈空泡状,含1~2个肥大的核仁,癌细胞间有淋巴细胞浸润
(4)未分化癌:癌细胞小而泡浆少,呈圆形或梭形,核圆形或卵圆形、浓染,细胞呈弥漫浸润,无明显癌巢形成。有时,癌组织主由短梭形细胞组成,颇似燕麦细胞癌(图9-7)。
图9-7 鼻咽未分化癌
癌细胞呈短梭形,核卵圆,浓染,呈燕麦细胞型癌结构
在鼻咽癌的组织学类型中,低分化鳞状细胞癌最为多见,其次为泡状核细胞癌,未分化癌和低分化癌较少,而高分化鳞状细胞癌和腺癌均极少见。
【扩展和转移】
(1)直接蔓延:癌组织向上蔓延可破坏颅底骨,并可经破裂孔侵入颅内,使第Ⅱ~Ⅵ对脑神经受损。向外侧扩展,可侵犯咽鼓管而进入中耳,引起耳症状。向前可侵犯鼻腔,甚至侵入眼眶。还可向后侵犯颈椎,甚至颈段脊髓。
(2)淋巴道转移:癌细胞常在早期就经淋巴道转移,先到咽后淋巴结,而后至颈深淋巴结上群。患者在乳突尖下方或胸锁乳突肌上段前缘出现无痛结节。颈淋巴结转移开始发生在同侧,继而发展为双侧。嗣后,颈侧中、下群淋巴结相继受累,并可互相融合形成巨大肿块。还可压迫第Ⅸ~Ⅻ对脑神经和颈交感神经引起相应症状。
(3)血道转移:多见于肝、肺和骨,也可转移至纵隔、硬脑膜、肾、肾上腺和胰等处。
【临床病理联系】
(1)鼻咽癌早期症状较复杂又往往不为患者重视,而且原发灶开始很小,不易发现,因而常被漏诊或误诊。只有做好肿瘤普查工作,警惕出现吸涕带血、耳鸣、鼻塞等症状,做细致的鼻咽部检查,方能早期发现,早期诊断。
(2)鼻咽粘膜淋巴管丰富,且其通透性很高,癌细胞常在早期就经淋巴道转移,颈淋巴结的转移率很高。常因同侧颈深淋巴结上群发生转移而在乳突尖下方或胸锁乳突肌上段前缘出现质硬的无痛结节。有60%以上的患者是以颈部肿块作为首发症状而就医,甚至有的病人颈淋巴结转移已由病理确诊多时,而临床上仍未能查出鼻咽部的原发癌灶。
(3)鼻咽癌组织因有迅速向深部浸润生长的倾向而破坏邻近组织,常侵犯颅底骨、咽鼓管、中耳、鼻腔及眼眶等处,还可侵入颅内,损害第Ⅱ~Ⅵ对脑神经,引起相应症状。
(4)鼻咽癌恶性程度较高,绝大多数为低分化鳞状细胞癌,泡状核细胞癌也颇常见,对放射治疗有高度或中度敏感性,但较易复发,尤以低分化鳞状细胞癌复发率较高。
(5)鼻咽癌组织的间质内炎性细胞反应情况与患者年龄、淋巴结转移、预后和血清中VCA-IgA水平有一定的关系。
第三节 喉部疾病
一、喉炎
(一)急性喉炎
急性喉炎(acute laryngitis)多发生于感冒之后,先有病毒入侵,再继发细菌感染所致。常见的致病菌有葡萄球菌、链球菌、肺炎球菌、流感杆菌等。此外,过多吸入生产性粉尘、有害气体(如氯、胺、熏烟、毒气等),烟酒过度,异物或检查器械损伤喉粘膜均可引起急性喉炎。由于病原体不同,其病变也各有特征。由感冒病毒引起者,多表现为急性卡他性喉炎,病变表现为粘膜充血、水肿和中性粒细胞浸润,并可出现粘液脓性分泌物。流感喉炎常表现为出血性炎症,加杂葡萄球菌和链球菌感染,可导致粘膜坏死和溃疡形成。白喉杆菌引起者表现为假膜坏死性喉炎。
(二)慢性喉炎
慢性喉炎(chronic laryngitis)可由急性喉炎逐渐演变而来,也可慢性潜隐开始。如吸入有害气体、化学粉尘、长期用声过度或发音不当,以及鼻、鼻窦、咽或下呼吸道感染均可成为喉部慢性刺激的来源。患者的主要症状是声嘶、喉部干燥、说话时感喉痛,常因喉部分泌物增加而觉有痰液粘附。病理变化有3种类型:①慢性单纯性喉炎:喉粘膜充血、红肿,镜下,粘膜及粘膜下组织充血、水肿,有一些淋巴细胞浸润,基底膜增宽、变疏松。②慢性肥厚性喉炎:喉头粘膜增厚,镜下,粘膜上皮增厚,可有鳞状上皮化生,并有角化现象;粘膜下纤维组织明显增生,有大量淋巴细胞、浆细胞浸润,甚至形成淋巴滤泡。③慢性萎缩性喉炎:喉粘膜萎缩变薄,干燥而发亮,声带变薄、张力减弱。镜下可见粘膜下纤维组织增生,粘膜萎缩和腺体萎缩、消失,纤毛柱状上皮被化生的鳞状上皮所代替。
声带小结(vocal nodules)是慢性喉炎的一种类型,多因长期用声过度或用声不当而致。小结多位于声带膜部的中心。初起时,小结柔软而带红色,其中血管扩张充血、间质水肿。随后,因发生纤维化和玻璃样变,表面的上皮棘层增厚,并有角化,小结因而变得坚实而呈苍白色。
(三)喉结核
喉结核(laryngeal tuberculosis)多由开放性肺结核经气道播散而来,原发于喉部的结核极少。其病变可分为3种类型:①浸润型:粘膜上皮增厚,固有膜中可见典型的结核结节,并有程度不等的纤维组织增生及淋巴细胞浸润。②溃疡型:以形成结核性溃疡为特征,溃疡面呈干酷样坏死,其下为结核性肉芽组织和纤维疤痕组织,溃疡附近的鳞状上皮常呈明显的乳头状增生或假上皮瘤样增生。③肿块型:结核病灶中纤维结缔组织明显增生形成肿块。结核结节常由大量纤维组织包绕,干酷样坏死常不明显。喉结核易蔓延至邻近组织,并经淋巴道播散引起颈淋巴结结核。
二、喉肿瘤
(一)良性肿瘤
喉乳头状瘤(papilloma of the larynx)是喉部良性肿瘤中最常见者。可发生于各年龄,但儿童和成人的喉乳头状瘤各有不同的特点。儿童喉乳头状瘤多发生于3~12岁儿童,可能由病毒感染引起,常在喉粘膜多处发生。镜下,呈典型乳头状瘤结构,电镜下,有时可检见细胞核内病毒包含体。被覆乳头的鳞状上皮角化不明显,很少发生恶性变。成人喉乳头状瘤常为单发性,往往位于声带的下侧面。被覆乳头之鳞状上皮每呈较显着的角化,较易发生癌变,故被视为癌前病变。
喉部的其他良性肿瘤,如腺瘤、血管瘤、纤维瘤、软骨瘤以及神经纤维瘤均较少见。
(二)恶性肿瘤
喉癌是较常见的上呼吸道恶性肿瘤,约占所有癌症的2%~3%。患者多在50~70岁之间,男多于女。常发生于吸烟较多者,此外,长期吸入有害物质、酗酒、环境污染以及喉乳头状瘤或喉粘膜白斑等,均可能与喉癌发生有关。喉癌可发生于喉内不同的部位,最多见者为声带癌,癌局限于声带内;其次是声门上癌,癌发生于声门上区,如会厌、杓会厌壁、室带和喉室等处;声门下癌较少,位于声带以下、环状软骨下缘以上的部位。喉癌的组织学类型中,鳞状细胞癌最多,约占95%~98%,腺癌较少,约占2%。喉癌常向粘膜下浸润生长,并可蔓延扩展侵犯附近软组织,亦可穿破环甲膜和破坏甲状软骨侵犯颈前软组织、甲状腺,癌向后扩散可累及食管,向下蔓延而至气管。喉癌转移一般发生较晚,常经淋巴道转移至颈部淋巴结,多见于颈总动脉分叉处淋巴结,然后沿颈内静脉向上、下部位的淋巴结扩展。血道转移可至肺、肝、肾和骨等处,但较少见。喉癌的转移与其发生部位、生长方式和分化程度有关。声带癌多分化较高、发展较慢,又由于声带淋巴管较少,因而转移率最低。声门上癌多分化较低、发展较快,加之该区淋巴管丰富,故颈淋巴结转移率最高。浸润型喉癌转移率高,而呈疣状生长的高分化鳞癌(喉疣状癌)则很少转移。
喉肉瘤很少见,约占喉肿瘤的1%,其中以纤维肉瘤较常见,横纹肌肉瘤、软骨肉瘤较少,此外,喉恶性淋巴瘤和恶性黑色素瘤极少。
第四节 气管支气管疾病
一、急性气管支气管炎
急性气管支气管炎(acute tracheo-bronchitis)是呼吸道常见疾病。在气候突变的季节容易发病,常继感冒、上呼吸道感染之后而发生,呼吸道局部防御功能削弱和全身抵抗力降低在发病中起重要作用。病毒(如鼻病毒、副流感病毒、腺病毒等)感染以及在病毒感染的基础上继发的细菌(如流感杆菌、肺炎链球菌等)感染常先引起上呼吸道炎症,并向下蔓延波及气管支气管粘膜,引起急性炎症。此外,吸入有害粉尘或气体(如二氧化硫、氯等)也可引起急性气管支气管炎。
【病理变化】
肉眼观,气管支气管粘膜红肿,表面粘附白色或带黄色的粘性分泌物,重症病例可见粘膜坏死和溃疡形成。组织学上,按其病变可分为:①急性卡他性气管支气管炎:粘膜呈斑状发红,表面覆有较稀薄的黄色粘性分泌物。若分泌物过多,造成支气管阻塞,则可引起通气障碍。②急性化脓性气管支气管炎:管壁呈化脓性炎症表现,炎症也可经细支气管管壁累及附近肺泡。患者常咳出黄色粘性脓痰。③溃疡性气管支气管炎:除感染因素外,也可因吸入有害气体、异物或胃内容物引起,大多仅发生表浅的粘膜坏死,坏死如较深则造成溃疡形成。炎症消退后,损伤程度轻者,粘膜上皮由粘膜基底层细胞增生而修复,气管支气管的结构和功能可恢复正常。损伤程度重者,管壁由疤痕修复,甚至引起支气管的疤痕性狭窄。
特殊类型的气管支气管炎包括白喉时的假膜性气管支气管炎和麻疹时的巨细胞性支气管炎等。
二、急性细支气管炎
急性细支气管炎(acute bronchiolitis)是指管径<2mm的细支气管的急性炎症。常见于4岁以下的婴幼儿,约90%的患儿在1岁以下。多在冬季发病,由病毒感染引起,主要是呼吸道合胞病毒,其次是腺病毒、副流感病毒,由腮腺炎病毒和流感病毒引起者较少。由于细支气管内腔狭窄,尤其是婴儿的小气道较成人更为狭窄,气流阻力增大,气流速度慢,故吸入的微生物易于沉积,加之,婴幼儿的特异性的非特异性免疫反应尚未成熟,支气管粘膜上的IgA水平较低,尚不能起保护作用,因而在感染呼吸道病毒后较成人更易患细支气管炎。由于细支气管内腔狭窄,管壁又无软骨支撑,发炎时易于阻塞或闭塞,因而患儿最突出的症状是喘憋性呼吸困难,严重者甚至可出现呼吸衰竭和窒息。
【病理变化】
细支气管粘膜肿胀,原呼吸上皮坏死脱落后,代之以增生的无纤毛柱状或扁平上皮,杯状细胞增多,粘液分泌增加,管壁内有淋巴细胞和单核细胞浸润。管腔内充满由纤维素、炎细胞和脱落的上皮细胞组成的渗出物,使管腔部分或完全阻塞,并可导致小灶性肺萎陷或急性阻塞性肺气肿。由于细支管管壁薄,炎症容易扩展累及周围的肺间质和肺泡,形成细支气管周围炎(peribronchiolitis)。如病变并不广泛,且其损伤程度不重,炎症消退后,渗出物被吸收或咳出而愈复。少数患者可因管壁的瘢痕修复,管腔内渗出物发生机化,使细支气管阻塞,形成纤维闭塞性细支气管炎(bronchiolitis fibrosa obliterans)。
三、支气管哮喘
支气管哮喘(bronchial asthma)是由于过敏反应或其他因素引起支气管弥漫性痉挛,出现发作性伴有哮鸣音的呼气性呼吸困难等典型症状的一种慢性疾病,也可视为慢性阻塞性支气管炎的一种特殊类型。
【病因和发病机制】
本病的病因较复杂,发病机制还未完全明了。目前一般认为,本病是在支气管反应性增高的基础上由于变应原或其他因素激发介质释放,引起支气管平滑肌痉挛性收缩所致。至于支气管反应性增高,则可能是与遗传有关的支气管β-肾上腺素受体封闭或敏感性降低所致。β-肾上腺素受体(腺苷环化酶)受刺激时,腺苷环化酶被激活,使细胞内ATP形成CAMP,从而抑制介质的形成和释放,使支气管舒张。当β-肾上腺素受体封闭或敏感性丧失时,支气管受刺激后易发生支气管痉挛。关于发病机制,目前公认的是本病属Ⅰ型变态反应。激发Ⅰ型变态反应性哮喘的变应原较多,如花粉、屋尘、尘螨、动物皮屑、霉菌、某些食品、药物、某些工业粉尘及气体等。当抗原进入机体后,诱发B细胞产生较多的IgE,并结合到靶细胞(气道粘膜内的肥大细胞)上,当再次接触同种抗原时,抗原与IgE发生桥联反应,催化肥大细胞膜上的花生四稀酸代谢过程,通过环氧化酶途径生成前列腺素和血栓素A2(TxA2);通过脂质氧化酶途径生成白细胞三稀(LTs),肥大细胞脱颗粒后还能释放组胺、5-羟色胺及嗜酸性粒细胞趋化肽等介质,引起弥漫性支气管平滑肌痉挛,粘液分泌增多以及管壁上嗜酸性粒细胞浸润。其他因素,如呼吸道感染、寒冷空气、刺激性气体和精神因素等亦可诱发哮喘发作。
【病理变化】
肉眼观,肺过度膨胀,柔软疏松而有弹性,支气管管腔内含有粘稠痰液和粘液栓,偶尔可有支气管扩张。镜下,可见粘膜上皮层中杯状细胞增多,粘液腺增生,粘膜的基底膜增厚并发生玻璃样变,管壁平滑肌肥大,粘膜下及肥厚的肌层内有嗜酸性粒细胞及单核细胞浸润。管腔内有粘液栓填塞。粘液栓中往往可见尖棱状Charcot-eyden结晶(嗜酸性粒细胞膜蛋白成分)及Curschmann螺旋,此乃崩解的细胞和粘液成分一起构成的螺旋状粘丝。
四、支气管扩张
支气管扩张(bronchiectasis)是指支气管的持久性扩张,多发生于肺段以下Ⅲ~Ⅳ级小支气管。病程多呈慢性经过,患者出现慢性咳嗽、咳大量脓痰或反复咯血等症状,支气碘油造影是临床确诊支气管扩张的重要检查方法。
【病因和发病机制】
支气管扩张的发病基础多为支气管壁的炎性损伤和支气管阻塞。炎症造成阻塞,阻塞又导致感染,互相影响,管壁的慢性炎症破坏了管壁的平滑肌、弹力纤维,甚至软骨,从而削弱了支气管管壁的支撑结构。当吸气和咳嗽时管内压增高并在胸腔负压的牵引下引起支气管扩张,而呼气时却又因管壁弹性削弱而不能充分回缩,久之,则逐渐形成支气管的持久性扩张。此外,支气管扩张患者血浆免疫球蛋白、类风湿因子及免疫复合物水平升高,肺组织内支气管相关淋巴组织显著增生,免疫组化检测证明,其中T细胞和B细胞均明显增多,表明在支气管扩张时局部体液免疫和细胞免疫应答增强,呈现超免疫状态。而且,在支气管上皮及增生的淋巴组织中,表达降钙素和5-羟色胺的神经内分泌细胞也显著增多。提示支气管扩张发病机制中有神经内分泌和免疫系统参与。少数先天性支气管扩张病例是因支气管壁发育障碍使管壁薄弱所致,部分病例还可伴有肺发育不全、胰腺囊性纤维化或其他畸形。Kartagener综合征就是支气管扩张、鼻窦炎和内脏转位三者并存,患者因粘液纤毛排送系统缺陷,易发生呼吸道感染而促进支气管扩张发生。
【病理变化】
肉眼观支气管扩张多发生于一个肺段,也可在双侧多个肺段发生。病变支气管可呈圆柱状或囊状扩张。扩张支气管、细支气管可连续延伸至胸膜下(图9-8),亦可呈节段性扩张。扩张支气管的数目多少不等,圆柱状和囊状扩张可同时并存,甚而使肺呈蜂窝状。扩张的管腔内常潴有黄绿色浓性或血性渗出物。有的管壁可见因粘膜肥厚而形成的纵行皱襞。周围肺组织常发生程度不等的肺萎陷、纤维化和肺气肿。先天性支气管扩张常呈多囊状。
镜下支气管壁呈慢性炎症改变并有不同程度的组织破坏,或仅见不完整的平滑肌、弹力纤维或软骨片,甚或完全消失。支气管粘膜常有鳞状上皮化生(图9-9)。有些区域支气管周围新生的小气道上皮增生。支气管周围的淋巴组织明显增生。支气管周围的纤维组织也增生,逐渐发生纤维化、瘢痕化。

图9-8 支气管扩张症
肺切面,可见多数支气管显著扩张
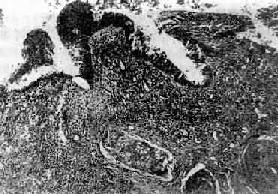
图9-9 支气管扩张症
支气管管壁呈现慢性炎症变化,粘膜上皮伴鳞状上皮化生
【合并症】
支气管扩张常因伴发化脓性感染而并发肺脓肿、脓胸、脓气胸。若经血道播散可引起脑膜炎、脑脓肿等。当肺广泛纤维化累及肺毛细血管床或形成支气管动脉与肺动脉分支吻合时,则可导致肺动脉高压,引起肺心病。此外,在支气管鳞状上皮化生基础上可发生鳞状细胞癌。
第五节 肺疾病
一、慢性阻塞性肺病
慢性阻塞性肺病(chronic obstructive pulmonary diseases,COPD)是一种慢性气道阻塞性疾病的统称,主要指具有不可逆性气道阻塞的慢性支气管炎和肺气肿两种疾病。
(一)慢性支气管炎
慢性支气管炎(chronic bronchitis)是40岁以上男性人群中最常见的疾病之一,临床上以反复发作咳嗽、咳痰或伴有喘息症状为特征,且症状每年持续约3个月,连续两年以上。病情进展,常常并发肺气肿和慢性肺源性心脏病。是一种严重影响健康的慢性病。
【病因和发病机制】
慢性支气管炎往往是因多种因素长期综合作用所致。起病与感冒有密切关系,多在气候变化比较剧烈的季节发病。呼吸道反复病毒感染和继发性细菌感染是导致慢性支气管炎病变发展和疾病加重的重要原因。吸烟与慢性支气管炎的关系也是肯定的,吸烟者比不吸烟者的患病率高2~8倍,吸烟时间愈久,日吸烟量愈大,患病率愈高,戒烟可使病情减轻。此外,长期接触工业粉尘、大气污染和过敏因素也常是引起慢性支气管炎的原因,而机体抵抗力降低,呼吸系统防御功能受损则是发病的内在因素。
【病理变化】
各级支气管均可受累,受累的细支气管愈多,病变愈重,后果也愈严重。主要的病变有:①粘膜上皮纤毛倒伏,甚至脱失。上皮细胞变性、坏死脱落。上皮再生时,杯状细胞增多,并可发生鳞状上皮化生(图9-10);②粘液腺肥大、增生,分泌亢进,浆液腺发生粘液化(图9-11);③管壁充血,淋巴细胞、浆细胞浸润;④管壁平滑肌束断裂、萎缩,而喘息型患者,平滑肌束可增生、肥大,管腔变窄;⑤软骨可发生变性、萎缩,钙化或骨化。支气管炎反复发作的结果,病变不仅逐渐加重,而且逐级向纵深发展蔓延,受累的细支气管数量也不断增多。细支气管因管壁薄,炎症易向管壁周围组织及肺泡扩展,导致细支气管周围炎,而且还可发生纤维闭塞性细支气管炎,是引起慢性阻塞性肺气肿的病变基础。

图9-10 慢性支气管炎
支气管粘膜纤毛上皮出现较多杯状细胞,部分呈磷状上皮化生

图9-11 慢性支气管炎
支气管粘膜上皮出现较多杯状细胞,固有层及粘膜下层内有慢性炎性细胞浸润,腺体呈粘液化
【临床病理联系】
患者因支气管粘膜的炎症和分泌物增多,而出现咳嗽、咳痰症状。痰一般呈白色粘液泡沫状。在急性发作期,咳嗽加重,并出现粘液脓性或脓性痰。由于支气管痉挛或支气管狭窄及粘液、渗出物阻塞而引起喘息。检查时,两肺可闻及哮鸣音、干湿啰音。有的患者因粘膜和腺体萎缩(慢性萎缩性支气管炎),分泌物减少,痰量减少甚或无痰。病变导致小气道狭窄或阻塞时,出现阻塞性通气障碍,表现为第1秒用力呼吸量和最大通气量明显降低,合併肺气肿时,肺残气量明显增多,肺总量也增大。
(二)肺气肿
肺气肿(pulmonary emphysema)是指呼吸细支气管以远的末梢肺组织因残气量增多而呈持久性扩张,并伴有肺泡间隔破坏,以致肺组织弹性减弱,容积增大的一种病理状态。在成人尸检例中,约50%可发现不同程度的肺气肿,其中约6.5%的患得因此病死亡。
【病因和发病机制】
肺气肿是支气管和肺疾病常见的併发症。与吸烟、空气污染、小气道感染、尘肺等关系密切,尤其是慢性阻塞性细支气管炎是引起肺气肿的重要原因。发病机制与下列因素有关:
1.阻塞性 通气障碍慢性细支气管炎时,由于小气道的狭窄、阻塞或塌陷,导致了阻塞性通气障碍,使肺泡内残气量增多,而且,细支气管周围的炎症,使肺泡壁破坏、弹性减弱,更影响到肺的排气能力,末梢肺组织则因残气量不断增多而发生扩张,肺泡孔扩大,肺泡间隔也断裂,扩张的肺泡互相融合形成气肿囊腔。
此外,细支气闭塞时,吸入的空气可经存在于细支气管和肺泡之间的Lambert孔进入闭塞远端的肺泡内(即肺泡侧流通气),而呼气时,Lambert孔闭合,空气不能排出,也是导致肺泡内储气量增多、肺泡内压增高的因素。
2.弹性蛋白酶增多、活性增高与肺气肿发生有关的内源性蛋白酶主要是中性粒细胞和单核细胞释放的弹性蛋白酶。此酶能降解肺组织中的弹性硬蛋白、结缔组织基质中的胶原和蛋白多糖,破坏肺泡壁结构。慢性支气管炎伴有肺感染、尤其是吸烟者,肺组织内渗出的中性粒细胞和单核细胞较多,可释放多量弹性蛋白酶。同时,中性粒细胞和单核细胞还可生成大量氧自由基,能氧化α1-抗胰蛋白酶活性中心的蛋氨酸使之失活。α1-抗胰蛋白酶乃弹性蛋白酶的抑制物,失活后则增强了弹性蛋白酶的损伤作用。
α1-抗胰蛋白酶由肝细胞产生,是一种分子量为45000~56000的糖蛋白,它能抑制蛋白酶、弹性蛋白酶、胶原酶等多种水解酶的活性。遗传性α1-抗胰蛋白酶缺乏是引起原发性肺气肿的原因,α1-抗胰蛋白酶缺乏的家族,肺气肿的发病率比一般人高15倍,主要是全腺泡型肺气肿。但是,在我国因遗传性α1-抗胰蛋白酶缺乏引起的原发性肺气肿非常罕见,并不重要。而最重要的也是最常见的是慢性阻塞性肺气肿(继发性肺气肿)。
【类型及其病变特点】
肺气肿病变发生在肺腺泡(acinus),即Ⅰ级呼吸细支气管所分布的肺组织范围内。属肺泡性肺气肿(alveolar emphysema)。根据病变的确切解剖部位及分布范围的不同可分为:
1.弥漫性肺气肿
(1)腺泡中央型肺气肿:腺泡中央型肺气肿(centriacinar emphysema)病变累及肺腺泡的中央部分,呼吸细支气管病变最明显,呈囊状扩张(图9-12)。而肺泡管、肺泡囊变化则不明显。
(2)全腺泡型肺气肿:全腺泡型肺气肿(panacinar emphysema)病变累及肺腺泡的各个部位,从终末呼吸细支气管直至肺泡囊和肺泡均呈弥漫性扩张,遍布于肺小叶内(图9-13)。如果肺泡间隔破坏较严重,气肿囊腔可融合成直径超过1cm的大囊泡,形成大泡性肺气肿(图9-14)。
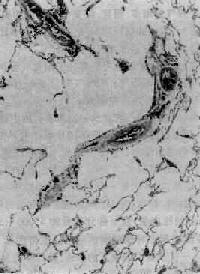
图9-12 腺泡中央型肺气肿
呼吸细支气管呈囊状扩张,伴行肺动脉(径80μm)管壁增厚,其分支内膜增厚,管腔极度狭窄

图9-13 全腺泡型肺气肿
末梢呼吸道弥漫性扩张,呈小囊状遍布于肺小叶内(径>200μm)
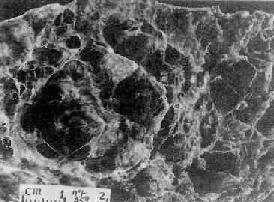
图9-14 左肺下叶大泡性肺气肿
在全腺泡型肺气肿基础上,胖有直径超过1cm的大囊泡
(3)腺泡周围型肺气肿:腺泡周围型肺气肿(periacinar emphysema)也称隔旁肺气肿(paraseptal emphysema),病变主要累及肺腺泡远端部位的肺泡囊,而近端部位的呼吸细支气管和肺泡管基本正常。常合并有腺泡中央型和全腺泡型肺气肿。
2.局限性肺气肿
(1)不规则型肺气肿:不规则型肺气肿(irregular emphysema)也称瘢痕旁肺气肿(paracicatricial emphysema),病变主要发生在瘢痕附近的肺组织,肺腺泡不规则受累,确切部位不定,一般是发生在呼吸细支气管远侧端,肺泡囊有时也受累。
(2)肺大泡:肺大泡(bullae of lung)病变特点是局灶性肺泡破坏,小叶间隔也遭破坏,往往形成直径超过2cm的大囊泡,常为单个孤立位于脏层胸膜下。而其余肺结构可正常。
间质性肺气肿(interstitial emphysema)是由于肺泡壁或细支气壁破裂,气体逸入肺间质内,在小叶间隔与肺膜连接处形成串珠状小气泡,呈网状分布于肺膜下。
【病理变化】
肉眼观:气肿肺显著膨大,边缘钝圆,色泽灰白,表面常可见肋骨压痕,肺组织柔软而弹性差,指压后的压痕不易消退,触之捻发音增强。
镜下:肺泡扩张,间隔变窄,肺泡孔扩大,肺泡间隔断裂,扩张的肺泡融合成较大的囊腔。肺毛细血管床明显减少,肺小动脉内膜呈纤维性增厚。小支气管和细支气管可见慢性炎症。腺泡中央型肺气肿的气肿囊泡为扩张的呼吸细支气管,在近端囊壁上常可见呼吸上皮(柱状或低柱状上皮)及平滑肌束的残迹。全腺泡型肺气肿的气肿囊泡主要是扩张变圆的肺泡管和肺泡囊,有时还可见到囊泡壁上残留的平滑肌束片断,在较大的气肿囊腔内有时还可见含有小血管的悬樑。
【临床病理联系】
肺气肿患者的主要症状是气短,轻者仅在体力劳动时发生,随着气肿程度加重,气短逐渐明显,甚至休息时也出现呼吸困难,并常感胸闷。每当合并呼吸道感染时,症状加重,并可出现缺氧、酸中毒等一系列症状。肺功能检查诊断肺气肿的标准是残气量超过肺总量的35%,最大通气量低于预计值的80%,肺总量超过预计值的100%,1秒用力呼吸量低于肺活量的60%。典型肺气中患者的胸廓前后径增大,呈桶状胸。胸廓呼吸运动减弱。叩诊呈过清音,心浊音界缩小或消失,肝浊音界下降。语音震颤减弱。听诊时呼吸音减弱,呼气延长,用力呼吸时两肺底部可闻及湿啰音和散在的干啰音。剑突下心音增强,肺动脉瓣第二音亢进。
肺气肿的严重后果有①肺源性心脏病及衰竭。②肺大泡破裂后引起自发性气胸,并可导致大面积肺萎陷。③呼吸衰竭及肺性脑病。由于外呼吸功能严重障碍,导致动脉血PaO2<8kPa(60mmHg),伴有或不伴有PaCO2<6.67kPa(50mmHg)的病理过程,称为呼吸衰竭(respiratory failure)。呼吸衰竭时发生的低氧血症和高碳酸血症会引起各系统的代谢功能严重紊乱。中枢神经系统对缺氧最为敏感,愈是高级部位敏感性愈高。随着缺氧程度的加重,可出现一系列中枢神系统功能障碍,由开始的大脑皮层兴奋性增高而后转入抑制状态。病人表现由烦燥不安、视力和智力的轻度减退,逐渐发展为定向和记忆障碍,精神错乱,嗜睡,惊厥以至意识丧失。迅速发生的CO2潴留也能引起中枢神经功能障碍,病人常出现头痛、头晕、烦燥不安、言语不清、扑翼样震颤、精神错乱以及嗜睡、昏迷、呼吸抑制等“二氧化碳麻醉”症状。由呼吸衰竭造成的以脑功能障碍为主要表现的综合征,称为肺性脑病(pulmonary encephalopathy),可能是由于低氧血症、高碳酸血症,以及酸碱平衡紊乱导致神经细胞变性、坏死和脑血液循环障碍引起脑血管扩张、脑水肿、灶性出血、颅内压升高甚至脑疝形成等因素综合作用所致。
二、肺炎
肺炎(pneumonia)通常是指肺的急性渗出性炎症,为呼吸系统的多发病、常见病。据世界卫生组织调查,肺炎死亡率占呼吸系统急性感染死亡率的75%。在我国,各种致死病因中,肺炎占第5位。肺炎可由不同的致病因子引起,根据病因可将肺炎分为感染性(如细菌性、病毒性、支原体性、真菌性和寄生虫性)肺炎,理化性(如放谢性、吸入性的类脂性)肺炎以及变态反应性(如过敏性和风湿性)肺炎。由于致病因子和机体反应性的不同,炎症发生的部位、累及范围和病变性质也往往不同。炎症发生于肺泡内者称肺泡性肺炎(大多数肺炎为肺泡性),累及肺间质者称间质性肺炎。病变范围以肺小叶为单位者称小叶性肺炎,累及肺段者称节段性肺炎,波及整个或多个大叶者称大叶性肺炎。按病变性质可分为浆液性、纤维素性、化脓性、出血性、干酪性、肉芽肿性或机化性肺炎等不同类型。
(一)细菌性肺炎
1.大叶性肺炎大叶性肺炎(lobar pneumonia)主要是由肺炎链球菌感染引起,病变起始于肺泡,并迅速扩展至整个或多个大叶的肺的纤维素性炎。多见于青壮年,临床表现为骤然起病、寒战高烧、胸痛、咳嗽、吐铁锈色痰、呼吸困难,并有肺实变体征及白细胞增高等。大约经5~10天,体温下降,症状消退。
【病因和发病机制】
95%以上的大叶性肺炎由肺炎链球菌引起,尤以Ⅲ型者毒力最强。此外,肺炎杆菌、金黄色葡萄球菌、溶血性链球菌、流感嗜血杆菌也可引起。受寒、疲劳、醉酒、感冒、麻醉、糖尿病、肝、肾疾病等均可为肺炎的诱因。此时,呼吸道的防御功能被削弱,机体抵抗力降低,易发生细菌感染。细菌侵入肺泡后在其中繁殖,特别是形成的浆液性渗出物又有利于细菌繁殖,并使细菌通过肺泡间孔或呼吸细支气管迅速向邻近肺组织蔓延,从而波及整个大叶,在大叶之间的蔓延则系带菌渗出液经叶支气管播散所致。
【病理变化】
病变一般发生在单侧肺,多见于左肺下叶,也可同时或先后发生于两个以上肺叶。病变基本特征是肺的微循环障碍。由于毛细血管通透性增高,大量纤维蛋白原渗出于肺泡,使肺组织大面积广泛实变。病变早期,肺叶充血、水肿,肺泡腔内有大量浆液性渗出物,混有少数红细胞、中性粒细胞和巨噬细胞,并含有大量细菌。1~2天后,即有大量纤维蛋白原渗出,肺泡腔内充满混有红细胞、中性粒细胞、巨噬细胞的纤维素性渗出物,纤维素丝可穿过肺泡间孔与相邻肺泡中的纤维素网相连(图9-15)。病变肺叶质实如肝,明显肿胀,重量增加,呈灰白色(图9-16)。如血管损伤较重、出血较多,外观可呈红色。大约经5~10天,炎症消退,细菌被吞噬细胞吞噬清除,渗出物被溶解,或经淋巴管吸收或被咳出。大叶性肺炎时,肺组织常无坏死,肺泡壁结构也未遭破坏,愈复后,肺组织可完全恢复其正常结构和功能。
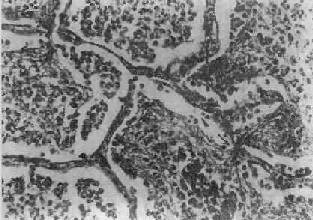
图9-15 大叶性肺炎
肺泡腔内充满纤维素性渗出物,纤维素丝穿过肺泡间孔,使相邻肺泡内的纤维素网互相连接

图9-16 大叶性肺炎
左下叶实变,呈灰白色,肺叶明显肿胀
【合并症】
(1)肺肉质变:因吞噬细胞数量少或功能缺陷,渗出物不能被完全吸收清除时,则由肉芽组织予以机化(图9-17),病变部位肺组织变成褐色肉样纤维组织,称肉质变(carnification)。
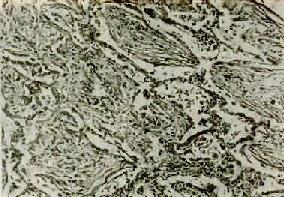
图9-17 肺肉质变
肺泡腔内炎性渗出物已被结缔组织所替代
(2)肺脓肿及脓胸或脓气胸:多见于由金黄色葡萄球菌引起的肺炎。
(3)纤维素性胸膜炎:是肺内炎症直接侵犯胸膜的结果。
(4)败血症或脓毒败血症:见于严重感染时,细菌侵入血流繁殖所致。
(5)感染性休克:严重的肺炎链球菌或金黄色葡萄球菌感染引起严重的毒血症时可发生休克,称休克型或中毒性肺炎,病死率较高。
【临床病理联系】
疾病发展过程中病变表现不一,临床体征也不相同。疾病早期时,主要病变是肺泡腔内浆液渗出,听诊可闻湿啰音,X线检查仅见肺纹理增深。肺实变时,由于肺泡膜面积减少,可出现肺泡通气和血流比例失调而影响换气功能,使肺静脉血不能充分氧合,患者乃出现紫绀或呼吸困难。渗出物中红细胞为肺泡巨噬细胞吞噬,崩解后形成含铁血黄素混入痰中,使痰呈铁锈色。痰中可检出“心衰细胞”。肺实变的体征是,肺泡呼吸音减弱或消失,出现支气管呼吸音,语音震颤增强,叩诊呈浊音。因常并发纤维素性胸膜炎,患者有胸痛,听诊可闻胸膜摩擦音。X线检查,可见大叶性或段性分布的均匀性密度增高阴影。病变消散时,渗出物溶解液化,肺部可闻及捻发音,X线表现为散在不均匀片状阴影,约在2~3周后阴影方完全消散。抗生素治疗,可缩短病程,减轻病变,合并症也大为减少。
2.小叶性肺炎小叶性肺炎(lobular pneumonia)主要由化脓菌感染引起,病变起始于细支气管,并向周围或末梢肺组织发展,形成以肺小叶为单位、呈灶状散布的肺化脓性炎。因其病变以支气管为中心故又称支气管肺炎(bronchopneumonia)。主要发生于小儿和年老体弱者。
【病因和发病机制】
小叶性肺炎主要由细菌感染引起,常见的致病菌有葡萄球菌、链球菌、肺炎球菌、流感嗜血杆菌、绿脓杆菌和大肠杆菌等。这些细菌通常是口腔或上呼吸道内致病力较弱的常驻寄生菌,往往在某些诱因影响下,如患传染病、营养不良、恶病质、慢性心力衰竭、昏迷、麻醉、手术后等,使机体抵抗力下降,呼吸系统的防御功能受损,细菌得以入侵、繁殖,发挥致病作用,引起支气管肺炎。因此,支气管肺炎常是某些疾病的并发症,如所谓麻疹后肺炎、手术后肺炎、吸入性肺炎、坠积性肺炎等等。
【病理变化】
小叶性肺炎的病变特征是肺组织内散布一些以细支气管为中心的化脓性炎症病灶。常散布于两肺各叶,尤以背侧和下叶病灶较多。病灶大小不等,直径多在1cm左右(相当于肺小叶范围),形状不规则,色暗红或带黄色(图9-18)。严重者,病灶互相融合甚或累及全叶,形成融合性支气管肺炎(confluent bronchopneumonia)。镜下,病灶中支气管、细支气及其周围的肺泡腔内流满脓性渗出物,纤维蛋白一般较少(图9-19)。病灶周围肺组织充血,可有浆液渗出、肺泡过度扩张等变化。由于病变发展阶段的不同,各病灶的病变表现和严重程度亦不一致。有些病灶完全化脓,支气管和肺组织遭破坏,而另一些病灶内则可仅见浆液性渗出,有的还停留于细支气管及其周围炎阶段。

图9-18 支气管肺炎
图中大小不等的不规则形区域即实变的支气管肺炎病灶
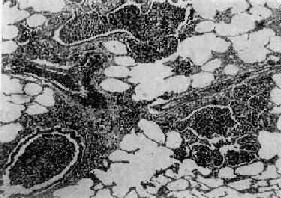
图9-19 支气管肺炎
图中见灶状实变的肺组织,肺泡内充满以中性粒细胞为主的炎性渗出物;病灶中有发炎的细支气管
【并发症】
小叶性肺炎发生并发症的危险性比大叶性肺炎大得多。可并发心力衰竭、呼吸衰竭、脓毒败血症、肺脓肿及脓胸等。支气管破坏较重且病程较长者,可导致支气管扩张。
【临床病理联系】
因小叶性肺炎多为其他疾病的并发症,其临床症状常为原发性疾病所掩盖。由于支气管粘膜的炎症刺激而引起咳嗽,痰呈粘液脓性。因病变常呈灶性散布,肺实变体征一般不明显。病变区细支管和肺泡内含有渗出物,听诊可闻湿啰音。X线检查,可见肺野内散在不规则小片状或斑点状模糊阴影。本病发现及时,治疗得当,肺内渗出物可完全吸收而痊愈。但在幼儿,年老体弱者,特别是并发于其他严重疾病时,预后大多不良。
(二)病毒性肺炎
病毒性肺炎(viral pneumonia)常常是因上呼吸道病毒感染向下蔓延所致。患者多为儿童,症状轻、重不等,但婴幼儿和老年患者病情较重。一般多为散发,偶可酿成流行。引起肺炎的病毒种类较多,常见的是流感病毒、还有呼吸道合胞病毒、腺病毒、副流感病毒、麻疹病毒、巨细胞病毒等等,也可由一种以上病毒混合感染并可继发细菌感染。病毒性肺炎的病情、病变类型及其严重程度常有很大差别。
【病理变化】
早期或轻型病毒性肺炎表现为间质性肺炎,炎症从支气管、细支气管开始,沿肺间质发展,支气管、细支气管壁及其周围、小叶间隔以及肺泡壁等肺间质充血、水肿,有一些淋巴细胞和单核细胞浸润,肺泡壁明显增宽(图9-20)。肺泡腔内一般无渗出物或仅有少量浆液。病变较重者,肺泡也可受累,出现由浆液、少量纤维蛋白、红细胞及巨噬细胞组成的炎性渗出物,甚至可发生组织坏死。有些病毒性肺炎(如流感病毒肺炎,麻疹病毒肺炎、腺病毒肺炎等)肺泡腔内渗出较明显,渗出物浓缩凝结成一层红染的膜样物贴附于肺泡内表面,即透明膜形成。支气管上皮的肺泡上皮也可增生,甚至形成多核巨细胞。麻疹病毒肺炎的病变特点为在间质性肺炎的基础上,肺泡壁上有透明膜形成,并有较多的多核巨细胞(巨细胞肺炎),在增生的上皮细胞和多核巨细胞的胞浆内和胞核内可检见病毒包含体。病毒包含体常呈球形,约红细胞大小,呈嗜酸性染色,均质或细颗粒状,其周围常有一清晰的透明晕。其他一些病毒性肺炎也可在增生的支气管上皮、支气管粘液腺上皮或肺泡上皮细胞内检见病毒包含体。如腺病毒肺炎可在增生的上皮细胞核内(图9-21),呼吸道合胞病毒肺炎可在增生的上皮细胞胞浆内,巨细胞病毒肺炎也可在增生的上皮细胞核内检见病毒包含体。检见包含体是病理组织学诊断病毒性肺炎的重要依据。
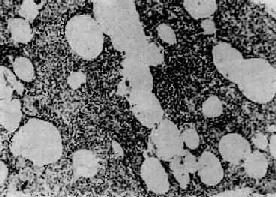
图9-20 间质性肺炎
肺泡壁及细支气管周围肺间质内有大量炎性细胞(主为单核细胞)浸润。肺泡壁明显增宽。肺泡腔内无渗出物
图9-21 腺病毒肺炎
图中央可见肿大肺泡上皮细胞中的核内包含体
有些混合感染,如麻疹病毒合并腺病毒感染,特别是又继发细菌感染的病毒性肺炎,病变更为严重,肺炎病灶可呈小叶性、节段性或大叶性分布。支气管和肺组织明显坏死、出血,并可混杂化脓性病变,从而掩盖了病毒性肺炎原来的病变特征。
(三)支原体肺炎
支原体肺炎(mycoplasmal pneumonia)是由肺炎支原体(mycoplasma pneumoniae)引起的一种间质性肺炎。支原体系介于细菌和病毒之间的微生物,共有30余种,其中多种可寄生于人体,但不致病,仅有肺炎支原体能引起呼吸道疾病。各种肺炎中约有5%~10%乃由肺炎支原体引起。主要经飞沫感染,秋、冬季节发病较多,儿童和青年发病率较高,通常为散发性,偶尔流行。患者起病较急,多有发热、头痛、咽痛及剧烈咳嗽(常为干性呛咳)等症状。胸部检查,可闻干、湿啰音。X线检查,肺部呈段性分布的纹理增加及网织状阴影。白细胞计数有轻度升高,淋巴细胞和单核细胞增多,痰、鼻分泌物及咽喉拭子能培养出肺炎支原体。
【病理变化】
肺炎支原体感染可引起整个呼吸道的炎症。肺部病变常仅累及一个肺叶,以下叶多见。病变主要发生于肺间质,病灶呈段性分布,暗红色,切面可有少量红色泡沫状液体溢出。气管或支气管腔内也可见粘液性渗出物。胸膜光滑。镜下,病变区域肺泡间隔明显增宽,有大量淋巴细胞、浆细胞和单核细胞浸润,肺泡腔内无渗出物或仅有少量混有单核细胞的浆液性渗出液。小支气管和细支气管壁及其周围组织也常有炎性细胞浸润。重症病例,上皮亦可坏死脱落,往往伴有中性粒细胞浸润。支原体肺炎预后良好。死亡率在0.1%~1%之间。
三、职业性肺疾病
在职业活动、特别是生产过程中,因长期吸入有害粉尘,引起以肺广泛纤维化为主要病变的疾病,统称尘肺(pneumoconiosis)。尘肺是我国一种法定职业病。属职业性尘肺的病种较多,按粉尘的化学性质可将其分为无机尘肺和有机尘肺两大类。无机尘肺中常见的有硅肺、煤工尘肺、石棉肺等。有机尘肺是因吸入各种有机尘埃,最常见的是由霉菌的代谢产物或动物性蛋白质引起的尘肺,如农民肺、蔗尘肺、蘑菇肺、麦芽肺和饲禽者肺等。尘肺对健康危害极大,关键在于预防。改革不合理的生产过程,建立粉尘监测制度,切实落实综合防尘措施。不接触粉尘或减少吸入粉尘的机会,对于粉尘作业工人定期体检,做到早期检查、早期诊断,对已确诊为尘肺患者及早调离粉尘作业,并进行必要的治疗,完全可以控制和减少尘肺的发病率。
(一)矽肺
矽肺(silicosis)是在生产环境中长期吸入大量含游离二氧化硅(SiO2)粉尘微粒所引起的以肺纤维化为主要病变的全身性疾病。游离二氧化硅主要存在于石英中,石英成分中SiO2占97%~99%。约有70%的矿石中均含有较多的SiO2。长期从事开矿、采石作业、坑道作业以及在石英粉厂、玻璃厂、耐火材料厂、陶瓷厂和搪瓷厂生产作业的工人易患本病。矽肺是危害最严重的一种职业病,其特点是发展缓慢,即使在脱离矽尘作业后,病变仍然继续缓慢发展。患者多在接触矽尘10~15年后才发病。若因吸入高浓度、高游离二氧化硅含量的矽尘,经1~2年后发病者,称速发型矽肺。矽肺的早期即有肺功能损害,但因肺的代偿能力很强,患者往往无症状,随着病变的发展,尤其是合并肺结核和肺心病时,则逐渐出现不同程度的呼吸和心功能障碍。
【病因和发病机制】
游离二氧化硅是矽肺的致病因子。矽肺的发生、发展与矽尘中游离二氧化硅的含量,生产环境中矽尘的浓度、分散度,从事矽尘作业的工龄及机体防御功能等因素有关。矽尘粒子愈小,分散度愈度,在空气中的沉降速度愈慢,被吸入的机会就愈多,致病作用亦愈强。一般来说,大于5μm的矽尘往往被阻留在上呼吸道,并可被呼吸道的防御装置清除。小于5μm的矽尘才能被吸入肺泡,并进入肺泡间隔,引起病变。尤以1~2μm的矽尘微粒引起的病变最为严重。
吸入肺泡内的矽尘微粒被肺巨噬细胞吞噬,沿肺淋巴流经细支气管周围、小血管周围、小叶间隔和胸膜再到达肺门淋巴结。当淋巴道阻塞后,矽尘沉积于肺间质内引起矽肺病变。若局部沉积的矽尘量多,引起肺巨噬细胞局灶性聚积,可导致矽结节形成;若矽尘散在分布,则引起弥漫性肺间质纤维化。矽肺的发病机制尚未完全阐明。一般认为,矽尘被肺巨噬细胞吞噬后,在游离二氧化硅的毒性作用下,巨噬细胞大量死亡崩解或发生功能和生物学行为改变,释放出一些致纤维化因子、包括巨噬细胞生长因子(MDGF),白细胞介素Ⅰ(IL-1)和纤维连结蛋白(FN)等,促进成纤维细胞增生和胶原形成,导致纤维化。至于巨噬细胞死亡的原因,主要是由于矽尘被巨噬细胞吞噬后,存在于次级溶酶体中,矽尘表层中的SiO2逐渐与水聚合成硅酸(系一种强的成氢键化合物),其羟基基团与溶酶体膜脂蛋白结构上的受氢原子(氧、氮或硫)间形成氢键,改变了溶酶体膜的脂质分子构型,从而破坏了膜的稳定性或完整性。溶酶体膜通透性增高或破裂,其中所含的大量水解酶溢出到细胞内,导致巨噬细胞自溶崩解。巨噬细胞死亡崩解后,释出的矽尘又被其它巨噬细胞吞噬,如此反复进行,使病变不断发展、加重。这也可解释何以患者脱离矽尘作业后肺部病变仍然会继续发展的缘由。
随着免疫学的发展,大量关于矽肺免疫的研究表明,在矽肺发生、发展过程中,有免疫因素参与。根据对矽结节玻璃样变组织的生化分析,其中球蛋白含量明显高于胶原含量,而有别于一般的玻璃样变组织的成分。动物实验证明,矽肺病变的纤维化程度与浆细胞反应强度呈正相关,提示矽肺的纤维化与抗原抗体反应有关。用荧光免疫组织化学方法观察矽结节,发现在胶原纤维及其间隙中有大量γ-球蛋白沉积,主要是IgG和IgM。如将尸检取得的矽结节玻璃样变组织制成匀浆,给家兔注射后,能产生抗人γ-球蛋白抗体。有人认为,浆细胞产生的免疫球蛋白通过形成抗原抗体复合物参与矽肺的发病。对矽肺患者作体液免疫测定发现,血清中IgG和IgM浓度增高,抗肺自身抗体、抗核抗体和类风湿因子检出率也较高。但关于矽肺免疫的抗原物质目前还未提取出来,多认为有3种可能性:①矽尘作为半抗原与机体的蛋白质结合构成复合抗原;②矽尘表面吸附的γ-球蛋白转化为自身抗原;③矽尘导致巨噬细胞死亡崩解后释放自身抗原。现已有很多证据表明,巨噬细胞死亡崩解后释放抗原的可能性最大。总之,矽肺的病因是明确的,发病机制极为复杂,在发病过程中可能有多种因素参与,它们互相影响、互为因果,共同促进矽肺的发生和发展。
【病理变化】
矽肺的基本病变是肺组织内矽结节形成和弥漫性间质纤维化。矽结节是矽肺的特征性病变,结节境界清楚,直径2~5mm,呈圆形或椭圆形,灰白色,质硬,触之有砂样感。随着病变的发展,结节可融合成团块状,在团块的中央,由于缺血、缺氧而发生坏死、液化,形成矽肺性空洞(silicotic cavity)。矽结节的形成过程大致分为三个阶段:①细胞性结节,由吞噬矽尘的巨噬结胞局灶性聚积而成,巨噬细胞间有网状纤维,这是早期的矽结节;②纤维性结节,由纤维母细胞、纤维细胞和胶原纤维构成;③玻璃样结节,玻璃样变从结节中央开始,逐渐向周围发展,往往在发生玻璃样变的结节周围又有新的纤维组织包绕。镜下,典型的矽结节是由呈同心圆状或旋涡状排列的、已发生玻璃样变的胶原纤维构成(图9-22)。结节中央往往可见内膜增厚的血管。用偏光显微镜观察,可以发现沉积在矽结节和肺组织内呈双屈光性的矽尘微粒。除矽结节外,肺内还有不同程度的弥漫性间质纤维化(图9-23),范围可达全肺2/3以上。此外,胸膜也因纤维组织弥漫增生而广泛增厚,在胸壁上也可形成胸膜胼胝,甚至可厚达1~2cm。肺门淋巴结内也有矽结节形成和弥漫性纤维化及钙化,淋巴结因而肿大、变硬。
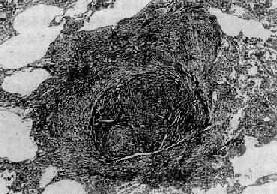
图9-22 矽肺
矽结节,由呈旋涡状排列的已发生玻璃样变的胶原纤维构成

图9-23 矽肺
肺组织呈弥漫性纤维化
矽尘沿血道转运,可在肝、脾、骨髓等处形成矽结节。
矽肺的分期和病变特征
根据肺内矽结节的数量、分布范围和直径大小,可将矽肺分为三期:
Ⅰ期矽肺:矽结节主要局限在淋巴系统。肺组织中矽结节数量较少,直径一般在1~3mm,主要分布在两肺中、下叶近肺门处。X线检查,肺野内可见一定数量的类圆形或不规则形小阴影,其分布范围不少于两个肺区。此时,肺的重量、体积和硬度无明显改变。胸膜上可有矽结节形成,但胸膜增厚不明显。
Ⅱ期矽肺:矽结节数量增多、体积增大,可散于全肺,但仍以肺门周围中、下肺叶较密集,总的病变范围不超过全肺的1/3。X线表现为肺野内有较多量直径不超过1cm的小阴影,分布范围不少于四个肺区。此时,肺的重量、体积和硬度均有增加,胸膜也增厚。
Ⅲ期矽肺(重症矽肺):矽结节密集融合成块。X线表现有大阴影出现,其长径不小于2cm,宽径不小于1cm,此时,肺的重量和硬度明显增加。解剖取出新鲜肺标本可竖立不倒(图9-24),切开时阻力甚大,并有砂粒感。浮沉试验,全肺入水下沉。团块状结节的中央可有矽肺空洞形成。结节之间的肺组织常有明显的灶周肺气肿,有时肺表面还可见到肺大泡。
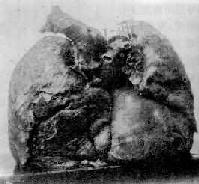
图9-24 Ⅲ期矽肺并发肺心病
两肺坚实能竖立,胸膜增厚。右心显著肥大、扩大,心尖由右心构成
【合并症】
1.矽肺结核病 矽肺合并结核病时称为矽肺结核病(silicotuberculosis)。愈是晚期、重症矽肺,肺结核的合并率愈高。Ⅲ期矽肺的合并率达60%~70%或更高。矽肺患者易合并肺结核可能是因游离二氧化硅对巨噬细胞的毒性损害以及肺间质弥漫性纤维化,导致肺的血液循环和淋巴循环障碍,从而降低了肺组织对结核杆菌的防御能力之故。矽肺结核病时,矽肺病变和结核病变可分开存在,也可混合存在(9-25)。矽肺结核病的病变比单纯矽肺和单纯肺结核的病变发展更快,累及范围更广,更易形成空洞。矽肺结核性空洞的特点是数目多,直径大,空洞壁极不规则。较大的血管易被侵蚀,可导致患者大咯血死亡。
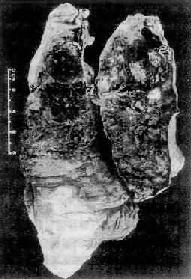
图9-25 矽肺结核病
图中可见胸膜增厚,切面上可见矽肺引起的肺气肿小囊泡。右侧白色斑块为结核病灶
2.肺感染由于矽肺患者抵抗力低,又有慢性阻塞性肺疾病,小气道引流不畅,故易继发细菌或病毒感染。尤其在有弥漫性肺气肿的情况下,肺感染可诱发呼吸衰竭而致死。
3.慢性肺源性心脏病 约有60%~75%的矽肺患者并发肺心病。这是因为肺间质弥漫性纤维化,肺毛细血管床减少,肺循环阻力增加。同时,矽结节内小血管常因闭塞性血管内膜炎,管壁纤维化,使管腔狭窄乃至闭塞,血管也扭曲、变形,尤以肺小动脉的损害更为明显,加之因呼吸功能障碍造成的缺氧,引起肺小动脉痉挛,均可导致肺循环阻力增加、肺动脉高压和右心室肌壁肥厚,心腔扩张。重症患者可因右心衰竭而死亡。
4.肺气肿和自发性气胸晚期矽肺患者常有不同程度的弥漫性肺气肿,主要是阻塞性肺气肿。有时,在脏层胸膜下还可出现肺大泡。气肿囊腔破裂引起自发性气胸。
(二)石棉肺
石棉肺(asbestosis)是因长期吸入石棉粉尘引起的以肺间质纤维化和胸膜肥厚为主要病变的疾病。石棉是一种天然的矿物结晶,其化学成分是含有铁、镁、铝、钙、镍等元素的硅酸盐复合物。石棉纤维具有耐酸、碱,隔热,绝缘等特性,在工业上用途很广。石棉矿的开采、选矿、运输工,石棉加工厂的分类、弹棉工,石棉制品的绝缘、隔热材料制品工等都可因长期吸入石棉粉尘而发生职业性石棉肺。发病工龄一般在10年左右。临床上,患者主要出现慢性支气管炎、肺气肿的症状,如咳嗽、咳痰、气急、胸胀痛等,晚期出现肺功能障碍和肺心病的症状和体征,患者痰内可检见石棉小体。
【发病机制】
吸入的石棉粉尘多数停留在呼吸性细支气管,仅有部分抵达肺泡,进入肺间质,有的还可被运输到脏层胸膜,引起肺间质和胸膜的结缔组织增生。至于纤维化形成的机制还不太清楚,可能主要是由于石棉纤维直接刺激纤维母细胞,促使脯氨酸羟化为羟脯氨酸,加速胶原合成,因而形成纤维化。除直接刺激作用外,也可能与石棉对巨噬细胞的毒性损害有关。虽然石棉对巨噬细胞的毒性作用比二氧化硅小,但石棉中的Mg2+对细胞膜也有溶解损伤作用。此外,石棉肺患者血清中IgM、IgG、抗核抗体和类风湿因子含量较高,肺内有异常球蛋白沉积,因而推测纤维化的形成,可能是巨噬细胞崩解,形成变性蛋白,引起自身免疫反应的结果。
【病理变化】
石棉肺的病变特点是肺间质弥漫性纤维化,其中可见石棉小体以及脏层胸膜肥厚和在壁层胸膜形成胸膜斑。因为吸入肺内的石棉纤维易随气流沿支气管长轴进入肺下叶,故肺病变以两肺下部为重,不同于矽肺病变以两肺中部为重的特点。
肉眼观,肺缩小、变硬,早期病变主要发生于两肺下部。由于细支气管周围、肺泡壁及小叶间隔内纤维组织增生,使肺呈明显的纤维网状结构。晚期,肺间质纤维化更广泛,更明显,肺组织陷于弥漫性纤维化。常因伴有明显的肺气肿和支气管扩张,肺组织可出现蜂窝状改变。肺胸膜尤其是下叶胸膜高度肥厚,晚期肺胸膜的纤维性增厚更广泛,甚至全肺均为灰白色的纤维瘢痕组织包裹。胸膜斑(pleural plaques)是指发生于壁层胸膜上的局限性纤维瘢痕斑块,境界清楚,凸出于胸壁,质地坚硬,呈灰白色、半透明而有光泽,状似软骨。胸膜斑常常位于两侧中、下胸壁,呈对称性分布。
镜下,早期病变为石棉粉尘引起的脱屑性间质性肺炎变化,表现为Ⅰ型肺泡上皮细胞受损,Ⅱ型肺泡上皮增生,后者呈立方形覆盖于肺泡表面。肺泡腔内有大量脱落的肺泡上皮细胞和巨噬细胞聚积。细支气管壁及其周围和血管周围的结缔组织中,以及肺泡间隔内有大量淋巴细胞和单核细胞浸润,有时也有一些嗜酸性粒细胞、浆细胞浸润。肺间质细胞增生,小动脉呈现闭塞性动脉内膜炎。随后,细支气管、血管周围及肺泡间隔内纤维结缔组织增生,到晚期则发展为肺组织弥漫性纤维化,大多数肺泡闭塞,由纤维组织填充。同时也可见一些衬以立方上皮的肺泡(腺样肺泡)。细支气管和小血管被包裹于纤维组织内,管壁纤维性增厚,管腔狭窄乃至闭塞。在增生的结缔组织纤维间散在着一些石棉小体(asbestos bodies),乃表面有铁蛋白沉积的石棉纤维,其长短不一(长者可超过100μm,短者仅数微米),黄褐色,呈哑铃状、分节状或蝌蚪形(图9-26),铁反应阳性。在石棉小体旁可见异物巨细胞。在弥漫性纤维化的肺组织中查见石棉小体是病理诊断石棉肺的重要依据。
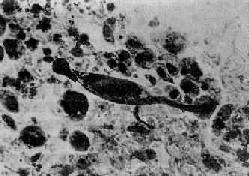
图9-26 石棉肺
图中央可见典型石棉小体,两端肥大,呈分节状,周围为尘细胞
【合并症】
动物实验和临床观察已证实,石棉属于促癌物质。近年来很多报道表明,石棉肺患者易并发肺癌、恶性胸膜和腹膜间皮瘤、食管癌、胃癌、结肠癌、喉癌等恶性肿瘤。据统计,石棉肺合并肺癌者可高达12%~17%,且多发于肺下叶。吸烟的石棉工人患肺癌的危险性比不吸烟人群高53~92倍。石棉肺合并恶性胸膜间皮瘤者也相当多见。有报告称,52例间皮瘤患者中约80%有接触石棉粉尘的职业史。从接触石棉粉尘到发现恶性间皮瘤的期限较长,平均42年。
石棉肺合并肺结核的频率约10%,远低于矽肺,而且多数病情较轻。此外,石棉肺易合并呼吸道感染、自发性肺气肿。晚期常并发肺源性心脏病和呼吸衰竭。
(三)农民肺
农民肺(farmer’s lung)是因吸入含有高温放线菌的有机粉尘而引起的一种外源性过敏性肺泡炎(extrinsic allergic alveolitis)。患者多系农业生产者,有经常接触发霉的稻草、麦秸和谷类等职业史。急性期时,典型病例在吸入含高温放线菌有机粉尘后4~8小时发病,出现畏寒、发热、呼吸困难或喘息、咳嗽、有少量粘液痰、乏力、全身肌肉酸痛、两肺可闻及湿啰音等症状和体征。急性期症状持续1~2天后可自行缓解,患者往往自认为是劳动后“受凉、感冒”,但也有的患者起病隐袭,呈现类似慢性支气管炎的症状。慢性期时,因反复发病,病变发展为弥慢性肺间质纤维化,并常并发肺心病,患者出现不同程度的心、肺功能障碍的症状和体征,重症患者可因呼吸衰竭和心力衰竭而死亡。农民肺的发病率,世界各地报告不同,一般为2.3%~8.6%。根据对湖北省洪湖县农民和城镇居民抽样调?
■[此处缺少一些内容]■
四、特发性肺间质纤维化
特发性肺间质纤维化(idiopathic pulmonary fibrosis,IPF)是指原因不明的弥漫性肺间质纤维化,为一种比较常见的肺疾病。可发生于任何年龄,患者以中、老年较多,多见于40~60岁之间。临床特征是出现进行性呼吸困难,X线显示两肺弥漫性网状结节状阴影,肺功能检查表明有限制性通气功能障碍、弥散功能障碍和肺的顺应性降低。病变特征是,早期表现为脱屑性间质性肺炎,晚期呈现不同程度的间质纤维化和蜂窝肺。多数患者呈慢性经过,但本病预后不佳,病死率甚高,常因肺功能不全和心力衰竭而死亡。平均生存期为5~6年,也有存活10年以上者。少数急性型病例进展急剧,多在6个月内死亡。年龄愈小者,病程愈短。
【病因和发病机制】
本病确切的病因和发病机制不明,可能与自身免疫、遗传因素和病毒感染有关。
目前较多的看法,认为特发性肺间质纤维化是一种自身免疫性疾病。其根据是本病常常与一些自身免疫性疾病,如类风湿性关节炎、硬皮病、系统性红斑狼疮等同时发生,而且在这些疾病中,肺部发生的病变与IPF的病变极其相似。IPF患者可出现高丙种球蛋白血症,升高的免疫球蛋白主要是IgG、IgM、IgA。部分病人可检出自身抗体,尤其是抗核抗体,类风湿因子阳性率较高。患者支气管肺泡灌洗液和血清中可检出免疫复合物(IgG-抗原复合物及IgM-C3b-抗原复合物),循环中免疫复合物也可在肺泡毛细血管壁内沉积。免疫复合物激活肺巨噬细胞释放趋化因子,引起肺组织内中性粒细胞、单核细胞和嗜酸性粒细胞浸润,而且这些细胞能产生氧自由基,还能分泌一些蛋白酶,如胶原酶、弹性硬蛋白酶等引起肺组织损伤,巨噬细胞还可产生纤维连接蛋白(fibronectin),促进纤维细胞增生,形成纤维化。
本病亦有家族性,有些患者是双胞胎,同家族的患者,蛋管异地居住多年,仍可发生同样的疾病。遗传连锁的研究表明,家族性IPF发生的危险与免疫球蛋白的γ异型有关。此外,在IPF患者中,存在HLA-B8、HLA-B12、HLA-B15和HLA-DW3、HLA-DW6、HLA-DR2抗原者增多,均提示本病发生与遗传因素有关。
约40%患者症状发作时有流感样表现及胸部症状,也有证据表明患者发病前有接触病毒史。有人报告,IPF患者血清中EB病毒抗体增加,可测出对病毒壳体抗原的IgA。提出EB病毒可能在IPF的病因学中起重要作用。
【病理变化】
本病早期,病变表现为脱屑性间质性肺炎,Ⅱ型肺泡上皮增生,肺泡腔内充满脱落的肺泡上皮细胞、肺泡巨噬细胞和淋巴细胞、中性粒细胞,有时可见透明膜形成。肺间质水肿,其中有灶性单核细胞、淋巴细胞和中性粒细胞浸润,以及广泛的纤维母细胞增生,肺泡间隔显著增宽。随着病情进展,肺泡腔和肺间质中的细胞逐渐减少。肺泡腔内炎性渗出物可发生机化,肺间质纤维组织逐渐增多,可发展至弥漫性间质纤维化。细支气管亦可见阻塞性细支气管炎病变,肺小动脉可呈现闭塞性动脉炎。细支气管和血管周围的纤维化也极为明显。到晚期,弥漫性纤维化使肺体积缩小、质地变硬,细支气管扩张呈小囊状,肺泡也不规则扩张成小气囊,小囊腔的囊壁破裂可形成较大的囊腔,这些囊被包裹于增生的纤维结缔组织之中,形成蜂窝肺。
【合并症】
特发性肺间质纤维化常合并肺部感染而导致呼吸衰竭。患者也常合并肺气肿、细支气管扩张和肺源性心脏病。部分患者可出现自发性气胸。少数病人可有肺性肥大性骨关节病。晚期特发性肺间质纤维化患者中,肺泡细胞癌、燕麦细胞癌、肺腺癌的发生率较高。
五、慢性肺源性心脏病
慢性肺源性心脏病(chronic cor pulmonale)是因慢性肺疾病引起肺循环阻力增加、肺动脉压力升高而招致的以右心室肥厚、扩张为特征的心脏病,简称肺心病。据流行病学调查,在我国肺心病的发病率较高,人群中的平均患病率为0.48%,尤以东北和华北地区较多,在各种器质性心脏病中,肺心病所占的百分比分别为18%~37%和12%~34%。
【病因和发病机制】
各种慢性肺疾病所致的肺循环阻力增加暨肺动脉高压是引起肺心病的关键环节。①慢性阻塞性肺疾病时,小气道的阻塞导致通气障碍,以及肺感染、肺间质纤维化及肺气肿均能破坏肺的血气屏障结构,减少气体交换面积,导致换气功能障碍。使肺泡气氧分压降低(缺氧),二氧化碳分压增高,引起肺小动脉痉挛(缺氧可干扰血管平滑肌细胞膜钾、钠离子交换和促使肥大细胞释放血管活性物质,引起肺小动脉痉挛)。②缺氧还能导致肺血管构型的改变,使肺小动脉中膜肥厚、无肌性细动脉的肌化,从而导致肺循环阻力增加和肺动脉高压。③限制性肺疾病,如胸廓病变、脊柱弯曲、胸膜纤维化及胸廓成形术后等,不仅可引起限制性通气障碍,还可压迫较大的肺血管和造成肺血管的扭曲,导致肺循环阻力增加暨肺动脉高压。④肺血管疾病,如反复的肺动脉栓塞和原发性肺血管疾病也可减少肺血管床面积而导致肺循环阻力增加和肺动脉高压。对增高的肺循环阻力,肺小动脉发生管壁平滑肌肌化增强,右心室也发生心肌细胞的适应性肥大,但右心室心肌细胞的适应能力是有限度的,当右心室负荷增高2~3.5倍时,极易出现心腔扩张。因此,肺心病可视为肺小动脉和右心室对慢性肺疾病引起的肺循环阻力和压力增高而发生的适应性反应,属于一种特殊的心脏病。
【病理变化】
1.肺部病变除原有的慢性支气管炎、肺气肿、肺间质纤维化等病变外,肺心病时肺内主要的病变是肺小动脉的变化,表现为肌型小动脉中膜肥厚、内膜下出现纵行肌束,无肌性细动脉肌化。还可发生肺小动脉炎,肺小动脉弹力纤维和胶原纤维增生以及肺小动脉血栓形成和机化。此外,肺泡壁毛细血管数量显著减少。
2.心脏病变右心室肥厚,心腔扩张,扩张的右心室使心脏的横径增大,并将左心室心尖区推向左后方,形成横位心,心尖主由右心室构成。心尖钝圆、肥厚。心脏重量增加(图9-27)。右心室前壁肺动脉圆锥显著膨隆(图9-28)。肥厚的右心室内乳头肌和肉柱显著增粗,室上嵴增粗,肺动脉圆锥处心肌壁增厚。通常以肺动脉瓣下2cm处右心室肌壁厚度超过5mm(正常约3~4mm)作为病理诊断肺心病的形态标准。镜下,可见心肌细胞肥大、增宽,核增大着色深。也可见缺氧所致的肌纤维萎缩、肌浆溶解、横纹消失,以及间质水肿和胶原纤维增生等现象。
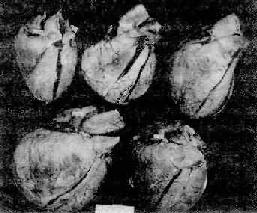
图9-27 正常与肺源性心脏病的心脏
图左上为正常成人心脏,其余为肺源性心脏病的心脏,其中最大者(左下)重785g
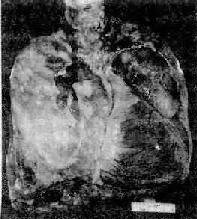
图9-28 慢性肺源性心脏病
两肺胸膜广泛性纤维性增厚,左肺上叶前段肺叶见肺大泡;左肺下叶被肥大转位的心脏压迫萎陷,右心室前壁肺动脉圆锥显著膨隆
【临床病理联系】
肺心病发展缓慢,临床表现除原有肺疾病的症状和体征外,主要是逐渐出现的呼吸功能不全和右心衰竭的症状和体征。受凉、上呼吸道感染、慢性支气管炎急性发作、肺炎及劳累等能诱发肺心病急性发作。每次急性发作都会进一步加重心、肺功能的损害,最后导致呼吸、循环衰竭。加强锻炼、增强体质,注意保暖并增强耐寒能力,戒烟,避开污染的空气,提高免疫力,预防诱发因素等对避免和减少肺心病的发生、发展至关重要。近年来,随着科学技术的进步,我国医务工作者正在从不同方面研究早期诊断肺心病的方法,如以X线检查、心电向量图和超声心动图检查,心、肺功能检查,放射性核素检查,肺阻抗血流图及肺微分阻抗血流图检查以及用漂浮导管技术直接测定肺动脉压力并进行血液动力学改变的研究和血液化学分析等,进行肺心病的早期诊断,将肺心病的防治工作提高到了新的水平。
六、肺不张和肺萎陷
肺不张(atelectasis)是指肺组织含气量过少,肺泡不能完全张开。肺萎陷(collapse of lungs)是指原已充满空气的肺组织因空气丧失而导致肺泡塌陷关闭的状态。
肺不张是先天性的,往往见于早产儿,由于呼吸中枢发育不成熟、分娩过程中引起的脑损伤或宫内窒息,使呼吸运动减弱或因细支气管被胎脂、胎粪、羊水阻塞,致出生时吸入的空气量不足而导致肺泡张开不全(新生儿肺不张)。肺发育不成熟,缺乏肺表面活性物质,使肺泡表面张力增加,肺的顺应性降低,也是导致新生儿肺不张的原因。肺萎陷是获得性的,多见于成人,有3种类型:①阻塞性(吸收性)肺萎陷:由于支气管被附近肿大的淋巴结或瘤块压迫,或支气管被肿瘤、异物、血凝块等阻塞,致空气不能进入所属末梢肺组织,其中原有的空气渐被吸收而导致一群小叶甚或整个肺叶无气和皱缩。②压缩性肺萎陷:大量胸腔积液,自发性气胸,严重的脊柱变形或腹腔巨大肿瘤或大量腹水使膈抬高,从而压迫肺组织,使肺泡陷闭而无气。③收缩性肺萎陷:由于肺组织广泛纤维化,纤维收缩所致。
七、呼吸窘迫综合征
(一)成人呼吸窘迫综合征
成人呼吸窘迫综合征(adult respiratory distress syndrome,ARDS)是由多种原因造成肺泡毛细血管膜弥漫性损伤和通透性增高而引起的一种急性低氧血症性呼吸衰竭。临床主要表现是患者突然发生进行性呼吸困难、紫绀、气促等急性呼吸窘迫症状,顽固性低氧血症和肺水肿,胸廓及肺的顺应性降低。X线检查显示迅速发展的两侧弥漫性肺浸润性阴影。成人呼吸窘迫综合征往往是严重创伤、大面积烧伤、大手术后、中毒、败血症或休克等患者严重的并发症。发病率有逐渐增高的趋势,病死率较高,据初步统计,其病死率为36%~68%。预后与肺病变的严重程度和肺功能状况有密切关系。
【病因和发病机制】
成人呼吸窘迫综合征是多种原因导致肺泡毛细血管膜(alveolocapillary membrane)急性损伤的结果。肺与外界相通,又是体内唯一接受全部心输出血量的器官,特别易受大气和血流中致病因子的伤害。而且,肺的血流量最多,毛细血管床的面积最大,受损的程度也比其他器官更为严重。致病原因如①肺感染;②吸入烟雾、毒气、胃内容物或溺水;③肺挫伤,放射性损伤;④氧中毒;⑤肺血栓栓塞,羊水栓塞,脂肪栓塞;⑥使用过量麻醉剂或某些药物(如水杨酸制剂、美散痛、海洛英、抗癌药等)以及⑦某些全身性病理过程(如败血症,脓毒败血症,休克,DIC,过敏反应,尿毒症,糖尿病酮症酸中毒,胰腺炎,严重创伤或烧伤等)和⑧某些治疗措施(如血液透析,体外循环,大量输出或输液)等都可引起肺泡毛细血管膜损伤,导致ARDS发生。其发病机制尚未完全阐明,可能主要与中性粒细胞和巨噬细胞在肺内聚积,释放大量蛋白水解酶(如胶原酶、弹性硬蛋白酶等)、氧自由基和一些血管活性物质(如前列腺素、白细胞三烯、血栓素A2等)引起肺泡毛细血管膜弥漫性损伤和通透性增高而导致肺水肿有关。此外,聚积在肺毛细血管内的血小板,既可阻塞毛细血管又可释放血管活性物质和溶酶体酶引起肺毛细血管膜损伤和通透性增高,在ARDS发病机制中起一定的作用。还有,肺表面活性物质减少,肺泡表面张力增加也是ARDS发病的因素之一。
【病理变化】
成人呼吸窘迫综合征的病理变化大致分为两个时期:
1.渗出期为病变早期,主要表现为肺间质毛细血管高度扩张、充血,肺间质和肺泡腔内有大量含蛋白质液体渗出(肺水肿)。在呼吸性细支气管及肺泡管或肺泡管与肺泡交界处的内表面有透明膜形成,其组成成分为蛋白质、纤维素和坏死的细胞碎屑。此外,肺间质中还可发生点状出血、灶性坏死以及淋巴管扩张。肺内往往发生局灶性肺萎陷。微血管内常有透明血栓和白细胞阻塞现象。
2.增殖期发病后数日,即转入损伤后的修复过程。Ⅱ型肺泡上皮细胞及肺间质的纤维母细胞大量增生,透明膜发生机化、纤维化,而且纤维化过程发展迅速。透明膜和肺泡间隔的纤维化很快扩展到全肺,导致弥漫性肺间质纤维化。
肉眼观,早期病变时肺表面湿润,有散在出血斑、出血点,并可见呈暗红色略凹陷的斑片状肺萎陷区。切面上,有大量泡沫状液体流出。3~4天后,肺出血、实变更加明显,呈牛肉样外观。可合并灶状梗死和支气管肺炎。
(二)新生儿呼吸窘迫综合征
新生儿呼吸窘迫综合征(neonatal respiratory distress syndrome,NRDS)指新生儿出生后已出现短暂(数分钟至数小时)的自然呼吸,继而发生呼吸过速、陷落呼吸(胸壁下陷)、呻吟、紫绀等急性呼吸窘迫症状和呼吸衰竭。患儿肺内形成透明膜为其主要病变特征,故又称肺透明膜病(hyaline membrane disease)。NRDS是新生儿发病率较高的疾病,主要见于妊娠不足35周龄的未成熟儿,糖尿病孕妇娩出的新生儿,体重过低的新生儿,剖腹产或臀位产的新生儿。此外,在第二个挛生儿也较常见。据报道,NRDS有家族性倾向,预后较差,病死率甚高。
【病因和发病机制】
新生儿呼吸窘迫综合征的病因与肺发育不全,缺乏肺表面活性物质有关。肺表面活性物质系由Ⅱ型肺泡上皮细胞合成和分泌,由磷脂酰胆碱、磷脂酰甘油等磷脂成分和蛋白质组成,具有降低肺泡表面张力,降低毛细血管内压和防止血浆滤出的作用。表面活性物质系统的发育至妊娠35周时才渐臻完善,其分泌达最高水平。肺发育不全,表面活性物质系统发育差(形态表现为Ⅱ型肺泡上皮细胞普遍缺乏板层小体),表面活性物质缺乏,使肺泡表面张力增加,肺顺应性降低,因而肺泡张开不全和不张。肺不张发生后,肺的通气和换气功能障碍导致缺氧、二氧化碳潴留和酸中毒,遂引起肺血管痉挛、肺血流灌注不足,从而损伤毛细血管内皮细胞,使毛细血管壁通透性增高,造成血浆蛋白漏出。同时,因缺乏表面活性物质,毛细血管内压增加,防止血浆滤出的作用降低,也促使血浆蛋白易于滤出。滤出到肺泡腔内的血浆蛋白凝集为透明膜,贴附于肺泡壁内表面,进一步影响气体交换,更加重了缺氧、二氧化碳潴留和酸中毒,肺血流灌注更为减少,进一步抑制肺表面活性物质的形成,如此形成恶性循环。
【病理变化】
肉眼观,肺含气少,色暗红,质韧,重量和体积可无明显改变。镜下,在呼吸性细支气管壁、肺泡管和肺泡壁上贴附一层均匀红染的膜状物(透明膜)。各肺叶均有不同程度的肺张开不全和不张,末梢气道中有水肿液。有些病例,肺间质和肺泡腔内出血较为明显。有的还可见吸入的羊水成分(鳞状上皮细胞和角化物质)。
八、肺癌
肺癌(lung cancer)是严重威胁生命的恶性肿瘤,为我国常见恶性肿瘤之一。半个世纪以来,世界许多国家和地区原发性肺癌的发病率和死亡率均有增加,尤以人口密度较高的工业城市更为突出。在欧美一些发达国家,肺癌的发病率和死亡率已升为各种癌症之首。在我国也有明显上升和继续上升的趋势。肺癌死亡率较高的地区主要在上海、天津、北京、辽宁、吉林、黑龙江等地。国内肺癌死亡率最高的城市是云南省个旧市,死亡率为70.62/10万。肺癌多发生于40岁以后,高峰发病年龄在70~79岁之间。男性患者多于女性,在我国男、女性比例为2.13:1。据西方国家统计,肺癌约占所有男性癌症的30%,而近年来,由于在西方国家女性吸烟者明显增多,女性肺癌患者的比例相应上升。
【病因】
肺癌的病因较为复杂,各国的研究表明,肺癌危险与下列因素有关:
1.吸烟国内外的大量研究证明,吸烟,尤其是嗜吸卷烟是引起肺癌的重要危险因素,吸烟者比不吸烟者的肺癌发生率高25倍,80%~90%的男性肺癌与吸烟有关。日吸烟量越大,开始吸烟的年龄越轻,患肺癌的危险越大。戒烟后患肺癌的危险性随戒烟时间的延长而逐渐降低。烟雾中含多种化学物质,如烟碱(居古丁)、多环芳烃碳氢化合物、镍、砷等与癌的发生有关,而细胞内的芳烃羟化酶(AHH)含量与吸烟者肺癌的发生呈正相关,肺癌患者芳烃羟化酶活性显著高于对照人群。苯并芘等多环芳烃碳氢化合物在芳烃羟化酶的作用下,转变为环氧化物,成为终致癌物,可与DNA共价结合,引起细胞的突变和恶性转化。
2.大气污染在大城市和工业区肺癌的发病率和死亡率较高,与大气污染有密切关系。工业和生活中能源(煤、柴油、汽油等)大量燃烧后的烟尘及产生的工业废气和生活废气是造成大气污染的重要原因。污染的空气中含有苯并芘、二乙基亚硝胺和胂等致癌物。许多国家的调查表明,工业城市中肺癌的死亡率与空气中苯并芘的浓度呈正相关。
3.职业因素肺癌的发生与某些职业有关。橡胶工人、镍业工人、石棉工人、铀矿、锡矿、荧石矿的采矿工人以及接触含砷粉制剂者肺癌的发生率很高,这与长期接触某种化学致癌物和放射性物质有关。如云南锡矿中肺癌发病率特别高,井下矿工肺癌发生率达到435.44/10万,可能与矿井中氡、氡子体和砷等因素有关。
4.电离辐射大剂量电离辐射可引起肺癌已为许多事实证明。人群中电离辐射的来源为自然界和医疗照射。铀矿和锡矿工人肺癌的危险是α射线造成的。日本原子弹伤害幸存者中肺癌明显增多。而且辐射的中子和α射线引起肺癌的危险性比单独α射线要高。还有报告指出,接受放射线治疗的患者肺癌的发生率也明显增高。
肺癌的组织发生
绝大多数肺癌均起源于各级支气管粘膜上皮,源于支气管腺体或肺泡上皮细胞者较少。因而肺癌实为支气管源性癌(bronchogenic carcinoma),包括鳞癌、腺癌、小细胞癌和大细胞癌几种主要类型。肺鳞癌主要起源于段和亚段支气管粘膜上皮,后者在致癌因子作用下,经鳞状化生、异型增生和原位癌等阶段再演进为浸润癌。肺腺癌来自支气管的腺体。细支气管肺泡细胞癌究系源于细支气管分泌粘液的上皮或富含糖原的Clara细胞,还是来自Ⅱ型肺泡上皮细胞,尚未最后定论。小细胞肺癌来源于位于支气管粘液腺和支气管粘膜内的Kultschitzky细胞(嗜银细胞)属APUD瘤。近年来有不少的报道,认为所有类型的肺癌均来自呼吸道粘膜的干细胞,它可向多方向分化,因而也可出现混合型癌。
【病理变化】
肉眼类型肺癌的肉眼形态多种多样,根据其部位和形态可分为3种主要类型:中央型、周围型和弥漫型。从尸检例看,中央型多于周围型,约为3:1。但从肺癌手术切除标本看,周围型则多于中央型,这是由于受手术指征限制的缘故。
(1)中央型:癌块位于肺门部,右肺多于左肺,上叶比中、下叶多见。癌由段支气管以上至总支气管发生,浸润管壁使管壁增厚、管腔狭窄甚或闭塞;进一步发展时,癌瘤沿支气管纵深方向浸润扩展,除浸润管壁外还累及周围肺组织,并经淋巴道蔓延至支气管肺淋巴结,在肺门部融合成环绕癌变支气管的巨大癌块(图9-29),形状不规则或呈分叶状,与肺组织的界限不清,有时比较清晰。癌块周围可有卫星灶。有时癌块内也可见坏死空腔。
图9-29 右主支气管中央型肺癌
主支气管管壁增厚,埋没于巨大分叶状癌块中
(2)周围型:癌发生在段以下的支气管,往往在近脏层胸膜的肺组织内形成直径2~8cm呈球形或结节状无包膜的癌块,与周围肺组织的界限较清晰,而与支气管的关系不明显(图9-30)。本型发生肺门淋巴结转移较中央型为迟,但可侵犯胸膜。Pancoast瘤是位于肺上叶顶部的肺癌,可由胸膜长入胸壁。

图9-30 右上叶周围型肺癌
肿块位于肺叶周边部,呈结节状,其境界较清晰,但与支气管的关系不明显
(3)弥漫型:此型罕见,癌组织沿肺泡管、肺泡弥漫性浸润生长,很快侵犯部分大叶或全肺叶,呈肺炎样外观,或呈大小不等的结节散布于多个肺叶内(图9-31)。此时须与肺转移癌和肺炎加以鉴别。

图9-31 弥漫型肺癌
肺内满布无数灰白色小癌结节
近年来,国内外对早期肺癌和隐性肺癌问题进行了不少研究,因为这对于肺癌的早期发现和早期诊断具有重要意义。有人主张,早期肺癌可分为管内型、管壁浸润型和管壁周围型三型,但无淋巴结转移。日本肺癌学会将癌块直径<2cm并局限于肺内的管内型和管壁浸润型列为早期肺癌。痰细胞学检查癌细胞阳性,而临床及X线检查阴性,手术切除标本经病理检查证实为原位癌或早期浸润癌而无淋巴结转移者为隐性肺癌。
组织学类型 肺癌的组织结构多种多样,根据1976年世界卫生组织所定,肺癌的组织学类型可分为鳞状细胞癌、腺癌、小细胞癌、大细胞癌4种基本类型。据统计,仅40%~50%病例呈一致性组织构型,其余则在肿瘤的不同部位表现不同分化状态的组织构型,其转移癌的组织学类型也可与原发癌不同。
(1)鳞状细胞癌:为肺癌中最常见的类型,约占50%~70%,尸检统计占35%~45%。患者以老年男性占绝大多数,多有吸烟史。肿块生长较慢,转移较迟。因其多来自段以上或总支气管,故较易被纤支镜检查发现,痰脱落细胞学检查阳性率最高,达88.25%。组织学上分为高分化、低分化和未分化三型。高分化鳞癌癌巢中多有角化珠形成,低分化鳞癌癌巢中仅有少量或无角化现象(图9-32),未分化型癌细胞多弥漫排列,癌巢不明显,核分裂像较多。三型中以低分化型居多。
图9-32 肺低分化鳞状细胞癌
癌巢主由具有间桥的癌细胞构成,无角化珠
(2)小细胞癌:发生率在肺癌中居第二位(临床统计在40%以上,尸检统计占15%~25%)。患者男多于女(20:1),发病年龄约在35~60岁。小细胞肺癌亦多发生于肺中央部,生长迅速,转移较早,恶生度高,5年存活率仅1%~2%。小细胞癌的癌细胞很小,呈短梭形或淋巴细胞样,有些细胞呈梭形或多角型,胞浆甚少,形似裸核。癌细胞常密集成群,由结缔组织加工分隔(图9-33)。有时癌细胞围绕小血管排列成假菊形团或管状结构。小细胞肺癌起源于支气管粘膜和粘液腺内Kultschitzky细胞,是一种具有异源性内分泌功能的肿瘤。
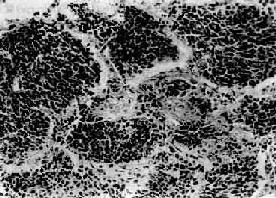
图9-33 小细胞肺癌
短梭形癌细胞平行排列,群集成团(燕麦细胞型)
(3)腺癌:发生率在肺癌中占第三位。临床统计占15%~20%,尸检统计占7%。患者女多于男(5:1),最常见于被动吸烟者。周围型肺癌中近60%为腺癌。肿块直径多在4cm以上,常累及胸膜,并常有肺门淋巴结转移。高分化腺癌癌巢呈腺管样结构(图9-34),可伴有粘液分泌;低分化腺癌的癌巢呈筛状或实体状;未分化腺癌的癌细胞呈高度异型性,可呈肉瘤样结构。
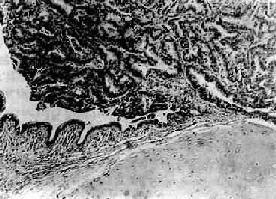
图9-34 肺高分化腺癌
腺癌自叶支气管粘膜发生,突入管腔内,癌组织主由腺样癌巢构成
肺腺癌的特殊类型还有腺样囊性癌、粘液癌、瘢痕癌和细支气管肺泡细胞癌等。瘢痕癌来自肺瘢痕组织,其中40%为腺癌,发展缓慢。细支气管肺泡细胞癌肉眼上呈弥漫型或多结节型。镜下,可见肺泡管和肺泡异常扩张,内壁衬以单层或多层柱状癌细胞,形成腺样结构,并常见乳头形成。而肺泡间隔大多保存完整。
(4)大细胞癌:癌细胞形成实体性癌巢或较大团块,主由胞浆丰富的大细胞组成,癌细胞高度异型。有时也可出现数量不等的多核癌巨细胞或胞浆空亮的透明细胞。此型恶性程度颇高,生长快,容易侵入血管形成广泛转移。
肺神经内分泌肿瘤(neuroendocrine tumors of lung)是指起源于APUD细胞,含有神经内分泌颗粒,能产生肽类激素的肺肿瘤,曾称为类癌。近20年来,随着电子显微镜和免疫组织化学技术的进展,虽然认识了肺神经内分泌肿瘤更多的形态和功能特征,但也带来了对该肿瘤诊断标准和分型的意见分歧,目前尚无统一意见。
1981年世界卫生组织将小细胞肺癌分为3个亚型:①燕麦细胞型:细胞呈短梭形或淋巴细胞样,②中间细胞型:细胞呈梭形或多角形,③混合型:小细胞癌伴有少量不典型鳞癌和分化不良的腺癌结构。1985年Palagudu将起源于Kultschitzky细胞的肺癌称为K细胞癌,并按其分化程度分为3个亚型:Ⅰ型:典型类癌,约有5.6%患者发生淋巴结转移,术后5年生存率高达95%;Ⅱ型:不典型类型,约有70%患者发生淋巴结转移或远处转移,术后5年生存率为65%;Ⅲ型:小细胞肺癌,是分化程度最低,恶性程度最高的一型,具有倍增时间(指肿瘤体积增大一倍所需的时间)短,浸润生长和广泛转移等特点,术后5年生存率<5%。各型肺神经内分泌肿瘤都具有表达神经肽类激素标志物的能力,表达的标志物可有蛙皮素、内皮素、胃泌素、促胃泌素释放肽、降钙素基因相关肽、血管活性肠肽等肽类激素和5-羟色胺、儿茶酚胺、乙酰胆碱等神经递质。此外,还含有神经元烯醇化酶和左旋多巴脱羧酶等物质。不同类型的肿瘤表达标志物的种类和数量虽有不同,但在同一种肿瘤中可有多种异源性肽类激素共存和肽类激素与神经递质共存的特点。鉴于肺神经内份泌肿瘤具有极其复杂的细胞表型和功能方面的异质性,而且应用电镜和免疫组化方法观察发现,肺神经内分泌肿瘤具有向APUD细胞和上皮细胞双向分化现象,特别是在小细胞肺癌中混杂的鳞癌和腺癌也可呈现上述各种标志物抗体免疫反应表达。提示不同组织学类型的肺癌可能有着共同的细胞起源。在同一肿瘤的细胞群体中出现形态和功能的异质性,可归因于体细胞异常转化的后代细胞所具有的多向性分化潜能。
【扩散途径】
1.直接蔓延 中央型肺癌常直接侵及纵隔、心包及周围血管,或沿支气管向同侧甚至对侧肺组织蔓延。周围型肺癌可直接侵犯胸膜,长入胸壁。
2.转移 肺癌发生转移较快、较多见。沿淋巴道转移时,首先至支气管肺淋巴结,再扩散至纵隔、锁骨上、腋窝、颈部淋巴结。血道转移常见于脑、肾上腺、骨以及肝、肾、胰、甲状腺和皮肤等处。
【临床病理联系】
肺癌的症状及其轻重与癌瘤的部位、大小和蔓延转移情况有关。肺癌一般发病隐匿,早期症状常不明显易被忽视。患者可有咳嗽、痰中带血、气急或胸痛。有时咯血,是最易引起注意而就医的症状。癌组织阻塞或压迫支气管时,可引起局限性肺萎陷或肺气肿。癌组织侵及胸膜可引起癌性胸腔积液;侵蚀食管可引起支气管食管瘘;侵犯纵隔内、气管旁淋巴结,压迫上腔静脉可引起上腔静脉综合征,表现为面颈部浮肿及颈、胸部静脉曲张。位于肺尖部的肺癌易侵犯交感神经链,引起病侧眼睑下垂,瞳孔缩小和胸壁皮肤无汗等交感神经麻痹综合征(Horner综合征);侵犯臂丛神经可出现上肢疼痛及手部肌肉萎缩。
肺癌,尤其是小细胞肺癌可有异位内分泌作用,可引起肺外症状。小细胞肺癌和腺癌可因5-羟色胺分泌过多而引起类癌综合征,表现为哮鸣样支气管痉挛,阵发性心动过速,水样腹泻,皮肤潮红等。肺性骨关节病(pulmonal osteoarthropathia)也是肺癌最常见的肺外症状,表现为伴有疼痛的骨、关节肥大和杵状指。此外,还可发生神经肌肉病变(肌无力综合征)、高血钙、低血糖和低钠血症、Cushing综合征及男性乳房发育症。这些肺外症状可在肿瘤切除后消失。所谓副肿瘤综合征(paraneoplastic syndrome)就是指除肿瘤及其转移灶直接引起的症状之外,伴随发生的由肿瘤引起的一系列异位激素性和代谢性症状综合征。肺癌、APUD瘤、胸腺瘤、肾透明细胞癌等都可引起副肿瘤综合征。
肺癌患者预后大多不良,早期发现、早期诊断和早期治疗至关重要。对于40岁以上的成人,特别有长期吸烟史并伴有咳嗽、痰中带血、气急、胸痛等症状者,或无痰干咳及与体位有关的刺激性呛咳的患者,必须提高警惕,及时进行X线、痰涂片细胞学(图9-35)和纤维支气管镜等检查,以及取活检组织作病理学检查,对肺癌的早期诊断具有重要价值。
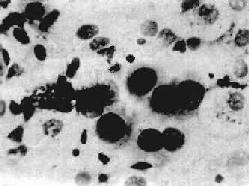
图9-35 痰涂片中的肺腺癌细胞
癌细胞圆形,核大、浓染,染色质粗,核仁大而明显,胞浆少
第十章 消化系统疾病
第一节 食管疾病
一、食管的狭窄与扩张
(一)食管狭窄
食管狭窄(stenosis of esophagus)有先天性狭窄及后天性狭二种。后天性狭窄可发生在以下三种病理情况:①食管粘膜上皮因炎症破坏或化学药品腐蚀,修复后形成瘢痕性狭窄。②食管肿瘤,多为食管癌,不同程度阻塞管腔。③来自食管周围组织的病变从外部压迫食管形成狭窄,如肺及纵隔的肿瘤、动脉瘤、甲状腺肿及淋巴结结核等。在狭窄部位的上方常见有食管的扩张及肥厚。
(二)贲门弛缓不能
贲门弛缓不能(achalasia)发生在食管的中下端及贲门。当食物通过时食管壁肌肉失去弛缓性调节而发生下咽困难(dysphagia)。食管中下段的管壁平滑肌运动功能受Auerbach神经丛调节。如该处神经节细胞发生器质性或功能性异常,甚至完全缺损时,则发生食管壁肌肉痉挛从而引起本病。由于中下段食管痉挛狭窄常伴发食管上段扩张,贲门部也发生痉挛,其肌层亦显肥厚。
(三)食管扩张
食管扩张(dilatation of esophagus)可分为原发性及继发性二种。发生在食管狭窄部上方的扩张属于继发性扩张,本节仅叙述原发性扩张,可分为广泛性扩张及限局性扩张两种。
1.广泛性扩张又称先天性扩张,发病原因不明,食管神经肌肉功能障碍引起全段食管扩张。由于食管全段扩张又称巨大食管症(megaesophagus)。
2.限局性扩张 食管的限局性扩张即憩室(diverticulum)常分为膨出性(真性)憩室及牵引性(假性)憩室二种。膨出性憩室常因食管壁平滑肌层先天发育不良,表面的粘膜部分由该处膨出(herniation)所致,多发生在咽、食管交界处,少数发生在食管下段。憩室多突出于后壁,由于憩室的增大在脊柱前方下垂;由于憩室内食物积存常压迫食管造成狭窄。牵引性憩室常因食管周围组织的慢性炎症(如淋巴结结核等)造成瘢痕性收缩,牵拉食管壁而形成,多发生在食管前壁,呈漏斗状扩张。
二、食管的炎症
1.急性食管炎常见的有单纯性卡他性及化脓性食管炎二种。卡他性者常因食入刺激性强的或高温食物引起。化脓性者多继发于食管憩室有食物潴留、腐败及细菌感染时,或形成脓肿,或沿食管壁扩散形成蜂窝织炎,进一步扩展可续发纵隔炎、胸膜炎及脓胸。
坏死性食管炎(necrotic esophagitis)见于某些传染病如白喉、猩红热等。白喉时多由咽喉部的粘膜坏死形成的假膜波及食管引起假膜性食管炎。此外,强酸、强碱等化学腐蚀剂引起的腐蚀性食管炎(corrosive esophagitis)常发生食管粘膜坏死及溃疡形成。愈合后可引起瘢痕狭窄。
2.慢性食管炎单纯性慢性食管炎常因习惯性摄食刺激性食物,重度吸烟,以及食管狭窄致食物潴留和慢性淤血等引起。此时由于慢性炎性刺激常有食管上皮限局性增生及不全角化,有时形成食管粘膜白斑(leukoplakia esophagi)。
反流性食管炎(regurgitant esophagitis)是由于胃液返流至食管,在食管下部引起的粘膜炎性病变。引起胃液返流的原因是各种因素引起的食管下段括约肌功能减弱,如呕吐、食管贲门切除术、胃内容潴留及置胃管等。反流胃液中的胃酸和胃蛋白酶可损伤食管粘膜使之发生充血、水肿及急性炎性细胞浸润。表层粘膜上皮细胞脱落形成糜烂乃至溃疡。晚期,溃疡处粘膜下层及肌层可有瘢痕形成,引起食管狭窄。
慢性反流性食管炎时食管下段粘膜的鳞状上皮可被胃粘膜柱状上皮所取代,称为Brrett食管,该处可发生溃疡或癌变。癌变率可达10%,多为腺癌。
Mallory-Weiss综合征由食管下部至贲门表面粘膜因损伤而发生龟裂并引起大出血。裂隙深度多达到粘膜下层。其发生原因多因暴饮暴食及其它原因引起的激烈呕吐。上述原因均可引起食管贲门处粘膜发生机械性裂伤。
三、食管癌
食管癌(carcinoma of esophagus)由食管粘膜上皮或腺体发生,占食管肿瘤的绝大多数。祖国医学称本病为噎隔。患者男多于女,发病年龄多在40岁以上,尤以60岁以上者居多。本病在我国华北及河南地区多发,高发区集中在太行山区附近,河南省林县是主要高发区。
【病因】
饮食因素在本病的病因中较为重要。曾认为饮酒、吸烟及食用过热饮食的习惯与本病的发生有关,但还有待深入研究。在我国高发区调查发现,当地某些粮食及食品中含有一定量的亚硝胺,其检出率比非高发区高。有些亚硝胺类化合物可以选择性诱发动物食管癌。此外也查出高发区居民食物常被真菌污染。用这种霉变食物能诱发大鼠前胃鳞状细胞癌。此外,也有认为患区地质土壤中缺钼等微量元素可能是引起食管癌的间接原因,其作用机制有待进一步研究。
【病变】
食管癌以食管中段最多见,下段次之,上段最少。可分为早期和中晚期两类。
1.早期癌此期临床上尚无明显症状。钡餐检查,食管基本正常或呈管壁轻度局限性僵硬。病变局限,多为原位癌或粘膜内癌,也有一部分病例癌组织可侵犯粘膜下层,但未侵犯肌层,无淋巴结转移。5年存活率在90%以上,预后较好。本型发现因难,发现率仅达6%左右。有可疑症状出现时,多通过食管拉网脱落细胞学检查,以检出癌细胞确诊。几乎全是鳞状细胞癌。
2.中晚期癌此期患者已出现临床症状,如吞咽困难等。肉眼形态可分为4型:
(1)髓质型:肿瘤在食管壁内浸润性生长,使食管壁均匀增厚,管腔变窄。切面癌组织为灰白色,质地较软似脑髓组织,表面可形成浅表溃疡。
(2)蕈伞型:肿瘤为卵圆形扁平肿块,如蘑茹状突入食管腔内。此型侵透肌层者较其他类型少见。
(3)溃疡型:肿瘤表面形成溃疡,溃疡外形不整,边缘隆起,底部凹凸不平,深达肌层(图10-1)。
(4)缩窄型:癌组织在食管壁内浸润生长,累及食管全周,形成明显的环形狭窄,近端食管腔明显扩张。
镜下,组织学上有鳞状细胞癌、腺癌、小细胞癌、腺棘皮癌等类型。其中以鳞状细胞癌最多见(图10-2),约占食管癌的90%,腺癌次之。大部腺癌来自贲门,极少数来自食管粘膜下腺体。近年偶有食管燕麦型小细胞癌的报告。
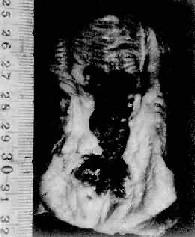
图10-1 食管癌
食管之中段,粘膜上见有一不规则的溃疡,边缘不整,底部不平。
图10-2 食管鳞状细胞癌
【扩散】
1.直接浸润癌组织穿透食管壁直接侵入邻近器官。食管上段癌可侵入喉部、气管和颈部软组织;中段癌多侵入支气管、肺;下段癌常侵入贲门、膈、心包等处。受浸润的器官可发生相应的合并症,如大出血、化脓性炎及脓肿、食管-支气管瘘等。
2.淋巴道转移转移沿食管淋巴引流途径进行。上段癌常转移到颈部及上纵隔淋巴结;中段癌多转移到食管旁及肺门淋巴结;下段癌常转移到食管旁、贲门及腹腔淋巴结。
3.血道转移 主要见于晚期患者,以转移到肝及肺为最常见。
食管其它恶性肿瘤
食管可发生平滑肌肉瘤、横纹肌肉瘤、恶性淋巴瘤等,但均甚少见。其总数约占食管恶性肿瘤的0.5%。此外,因正常食管的4%~8%存在黑色素母细胞,由此可发生恶性黑色素瘤,但其发生率远较皮肤等其他部位低。
第二节 胃肠疾病
一、胃炎
胃炎(gastritis)是胃粘膜的炎性变化,是一种常见病,可分为急性胃炎和慢性胃炎。慢性胃炎可由急性胃炎反复发作迁延而来,但多数因其他全身性因素及胆汁反流至胃内引起。近年发现,从慢性胃炎患者胃粘膜中分离出一种螺旋状弯曲杆菌,命名为弯曲样细菌(campylobacter like organisms,CLO),经研究证明本菌与胃炎之间关系密切,该菌不仅适应胃内高酸环境,还可降解胃粘膜表面被覆粘液使其失去保护作用,故被认为是慢性胃炎的一种病原因素。
(一)急性胃炎
急性胃炎(acute gastritis)常见者有4种:
1.急性卡他性胃炎(acute catarrhal gastritis)多因暴饮暴食引起,亦称刺激性胃炎(irritation gastritis)。胃粘膜充血、水肿有时可见粘膜糜烂。
2.腐蚀性胃炎(corrosive gastritis)由咽下高浓度酸、碱或腐蚀性化学剂引起。病变多较严重,胃粘膜出现坏死、软化溶解。
3.急性蜂窝织炎性胃炎(acute phlegmonous gastritis)少见,是一种弥漫性化脓性炎,病情较重。可由金黄色葡萄球菌、链球菌或大肠杆菌等化脓菌经血行感染(败血症)或直接感染(创伤)侵入胃壁引起。
4.急性出血性胃炎(acute hemorrhagic gastritis)主要表现为胃粘膜急性出血合并轻度坏死。本病发生与服用某些药物(如水杨酸制剂)、过量应用肾上腺皮质激素及过度饮酒有关。
(二)慢性胃炎
慢性胃炎(chronic gastritis)是一种常见病。一般分为表浅性及萎缩性二种。
1.慢性表浅性胃炎(chronic superficial gastritis)本病在胃窦部最为常见,为常见的胃粘膜疾患之一,纤维胃镜检出率高达20%~40%。
【病变】
肉眼观,病变多为多灶性或弥漫性,病变胃粘膜充血、水肿,有时并伴有点状出血或糜烂。镜下,可见炎性病变主要限于粘膜浅层,呈弥漫性或灶状分布。浸润的炎性细胞主要是淋巴细胞和浆细胞,有时可见少量嗜酸性粒细胞及中性粒细胞。此外可见粘膜浅层出现水肿、小出血点或表浅上皮坏死脱落。
2.慢性萎缩性胃炎(chronic atrophic gastritis)本病炎症改变并不明显,主要是胃粘膜的萎缩性变化。此时胃粘膜萎缩变薄,腺体减少或消失。临床上可有胃内游离盐酸减少或缺乏、消化不良,上腹不适或钝痛、贫血等症状。
【病因和分型】
现今将萎缩性胃炎分为A、B二型。已知A型发病与免疫因素关系密切,病人血中可找到抗胃壁细胞微粒体的自身抗体。病变在胃体部,并有维生素B12吸收障碍,常合并恶性贫血,可找到抗内因子抗体。B型的发病则与自身免疫无关,同时并不伴有恶性贫血也找不出自身抗体。其病因可能与吸烟、酗酒或滥用水杨酸类药物(如APC)等有关。B型亦称单纯性萎缩性胃炎,在我国较为多见。其病变部位在胃窦部,有的并可能发生癌变。两型萎缩性胃炎的胃粘膜病变基本一致。
【病变】
胃粘膜薄而平滑,皱襞变浅,有的几乎消失。粘膜表面呈细颗粒状。胃镜检查,可有3个特点:①正常胃粘膜的橘红色色泽消失,代之以灰色;②萎缩的胃粘膜明显变薄,与周围的正常胃粘膜界限明显;③萎缩处因粘膜变薄,粘膜下血管分支清晰可见。
镜下,病变区腺上皮萎缩,腺体变小并可有囊性扩张,常出现上皮化生(假幽门腺化生及肠上皮化生)。在粘膜固有层有不同程度的淋巴细胞和浆细胞浸润。在胃体和胃底部病变区,主要呈现壁细胞消失,其次是主细胞消失和粘液分泌细胞化生。后者称为幽门腺或假幽门腺化生。电镜观察,壁细胞除数量减少外,可见细胞内自噬泡增多,内质网扩张,高尔基器减少,线粒体肿胀,表面微绒毛消失。内质网、高尔基器和微绒毛是壁细胞的功能部分,其减少或消失反映了细胞泌酸功能的低下或消失。在幽门窦病变区,主要改变为幽门腺呈不同程度的萎缩、消失(图10-3)并常有肠上皮化生。胃粘膜表层上皮细胞生长,可形成绒毛样突起,增生的上皮中出现分泌粘液的杯状细胞(图10-4)、具有刷状缘的吸收上皮细胞和Paneth细胞时其形态结构与小肠粘膜相似,故称为肠上皮化生。现知肠上皮化生的胃粘膜易诱发胃癌,多为息肉样腺癌。
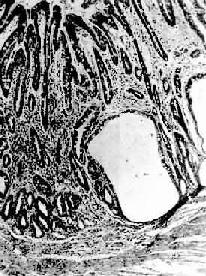
图10-3 慢性萎缩性胃炎
幽门腺大部分萎缩消失,胃小凹延长,有潴留性小囊形成,腺上皮中杂有不少杯状细胞(肠上皮化生),固有膜内有不少慢性炎性细胞浸润(Ⅱ74-4950)
胃粘膜的肠上皮化生分为小肠型及大肠型二种,二型中又分别分出完全肠上皮化生与不全肠上皮化生(图10-4)。小肠型与大肠型肠上皮化生可从光镜的腺体形态及粘液组化反应加以鉴别。Teglbjaerg曾报告含氧乙酰化唾液酸粘液者为大肠型化生,该粘液反应阴性者为小肠型化生。多数研究指出大肠型化生与肠型胃癌并存者较多,特别是不全大肠型肠上皮化生在癌旁组织中明显增多,其DNA含量及CEA的检出率均明显增加。但肠上皮化生究竟是否属于癌前病变迄今尚无定论。
图10-4 胃粘膜肠上皮化生
胃粘膜上皮出现多量充满粘液的杯状细胞
3.肥厚性胃炎(hypertrophic gastritis) 又称肥厚性胃病(hypertrophic gastropathy)、Menetrier病。发病原因不明。病变特点是,粘膜肥厚,皱襞加深变宽似脑回状(图10-5)。镜下,腺体肥大增生,腺管延长。粘膜固有层炎性细胞浸润不显著。粘膜表面粘液分泌细胞数量增加,壁细胞及主细胞有时减少。临床上,多数患者有胃酸低下及因蛋白由胃液丧失而致的低蛋白血症。
图10-5 慢性肥厚性胃炎
胃粘膜肥厚,皱襞加深变宽似脑回状
4.疣状胃炎(gastritis verrucosa)是一种有特征性病理变化的胃炎,病变处胃粘膜发生一些大小不等的糜烂,其周围粘膜隆起,因而形成中心凹陷的病灶,形如痘疹。病灶主要分布在幽门窦部。病变活动期,镜下可见病灶中心凹陷部的胃粘膜上皮处于变性、坏死和脱落状态并伴有急性炎性渗出物覆盖病灶表面。病变修复时局部粘膜上皮再生修复。有时可见修复上皮呈不典型增生。本病与一般的单纯性胃粘膜糜烂不同,其发病机制尚有待查清。
二、溃疡病
溃疡病亦称慢性消化性溃疡(chronic peptic ulcer)是常见病之一,多见于成人。患者有周期性上腹部疼痛、返酸、嗳气等症状。本病易反复发作,呈慢性经过。有胃及十二指肠溃疡2种。十二指肠溃疡较胃溃疡多见,据统计前者约占70%,后者约占25%,两者并存的复合性溃疡约占5%。
【病变】
胃溃疡多位于胃小弯,愈近幽门处愈多见,尤多见于胃窦部(图10-6)。在胃底及大弯侧十分罕见。溃疡通常只一个,呈圆形或椭圆形,直径多在2.5cm以内(图10-7)。溃疡边缘整齐,状如刀切,底部通常穿越粘膜下层,深达肌层甚至浆膜层。溃疡处粘膜下层至肌层可完全被侵蚀破坏,代之以肉芽组织及瘢痕组织(图10-8)。
图10-6 慢性胃溃疡好发部位示意图

图10-7 慢性胃溃疡
在胃小弯近幽门部有一直径约1cm的溃疡,边缘整齐,底部平坦,溃疡底部见一动脉裂口

图10-8 慢性胃溃疡模式图
溃疡略似斜置漏斗状,深达肌层
镜下,溃疡底大致由4层组织组成:最上层由少量炎性渗出物(白细胞、纤维素等)覆盖;其下为一层坏死组织;再下则见较新鲜的肉芽组织层;最下层则由肉芽组织移行为陈旧瘢痕组织(图10-9)。位于瘢痕组织内的小动脉因炎性刺激常有增殖性动脉内膜炎,使小动脉管壁增厚,管腔狭窄或有血栓形成。此种血管变化可引起局部血液循环障碍,妨碍组织再生使溃疡不易愈合。但这种变化却可防止溃疡底血管破溃、出血。另外,在溃疡边缘常可看到粘膜肌层与肌层粘连、愈着。溃疡底部的神经节细胞及神经纤维常发生变性和断裂。有时神经纤维断端呈小球状增生,这种变化可能是患者产生疼痛症状的原因之一。

图10-9 慢性胃溃疡底部
表层为炎性渗出物,其下方为坏死层,再下为肉芽组织及瘢痕组织
十二指肠溃疡的形态与胃溃疡相似,发生部位多在十二指肠起始部(球部),以紧接幽门环的前壁或后壁最为多见。溃疡一般较胃溃疡小而浅,直径多在1cm以内。
【结局及合并症】
1.愈合如果溃疡不再发展,渗出物及坏死组织逐渐被吸收、排除。已被破坏的肌层不能再生,底部的肉芽组织增生形成瘢痕组织充填修复,同时周围的粘膜上皮再生,覆盖溃疡面而愈合。
2.幽门狭窄 约发生于3%的患者。经久的溃疡易形成大量瘢痕。由于瘢痕收缩可引起幽门狭窄,使胃内容通过困难,继发胃扩张,患者出现反复呕吐。
3.穿孔 约见于5%的患者。十二指肠溃疡因肠壁较薄更易发生穿孔。穿孔后由于胃肠内容漏入腹腔而引起腹膜炎。
4.出血因溃疡底部毛细血管破坏,溃疡面常有少量出血。此时患者大便内常可查出潜血,重者出现黑便,有时伴有呕血。溃疡底较大血管被腐蚀破裂则引起大出血,约占患者的10%~35%。
5.癌变多见于胃溃疡,十二指肠溃疡几乎不发生癌变。癌变多发生于长期胃溃疡病患者,癌变率仅在1%或1%以下。癌变来自溃疡边缘的粘膜上皮或腺体,因不断受到破坏及反复再生,在此过程中在某种致癌因素作用下细胞发生癌变。
【病因和发病机制】
溃疡病的病因很复杂,尚未清楚。一般认为与以下因素有关:
1.胃液的消化作用多年研究已证明,溃疡的形成是胃壁或十二指肠壁组织被胃酸和胃蛋白酶消化的结果。这种自我消化过程是溃疡形成的直接原因。空肠及回肠内为碱性环境,极少发生这种溃疡。但作过胃空肠吻合术后,吻合处的空肠则可因胃液的消化作用而形成溃疡。
Zollinger-Ellison综合征时患者患有胰岛腺瘤,此瘤被认为来自胰岛D细胞。此瘤能分泌胃泌素样物质,使胃酸分泌极高,可达正常的10~20倍,故本瘤亦称为胃泌素瘤(gastrinoma),有时也见于十二指肠。由于胃泌素的大量分泌使胃、十二指肠,甚至空肠发生多数溃疡。已证明溃疡是由胃液消化作用所致。
十二指肠溃疡时还可见分泌胃酸的壁细胞总数增多,近正常的1倍。由此造成胃酸分泌增加,在空腹时尤甚。
正常情况下,胃粘膜不会被胃液消化,这是因为粘膜具有防御屏障功能:胃粘膜分泌的粘液,在胃粘膜表面形成一层粘液膜,覆盖于粘膜面,可以避免或减少胃酸直接接触粘膜,同时粘液对胃酸尚有中和作用。当胃粘膜的这种屏障功能受到损害时,分泌至胃腔内的胃酸中的氢离子得以弥散进入胃粘膜(逆向弥散)。氢离子由胃腔进入胃粘膜的弥散能力在胃窦部为胃底的15倍,而十二指肠又为胃窦的2~3倍。溃疡好发于十二指肠及胃窦部可能与此有关。由于胃酸氢离子的逆向弥漫,在临床测定胃溃疡患者胃内的酸度时,有时反而出现正常或过低的现象。此外,胆汁可以改变胃粘膜表面粘液层的特性,从而损害胃粘膜屏障功能。所以,十二指肠内容物特别是胆汁反流入胃在胃溃疡的发生上有着重要作用。
2.神经、内分泌功能失调十二指肠溃疡病患者胃酸分泌增多的原因与迷走神经的过度兴奋有关。正常胃酸分泌有二种时相:一为通过迷走神经对胃粘膜壁细胞的胆碱能性刺激引起胃泌素的释放(脑相,cephalic phase);另一为食物在胃内滞留直接刺激胃窦部粘膜引起胃泌素的释放(窦相,antral phase)。十二指肠溃疡病患者在空腹时,由于迷走神经功能亢进也经常有胃液分泌的增加,这是十二指肠溃疡病的特点。胃溃疡则与此不同。此时,迷走神经的兴奋性反而降低,致胃蠕动减弱,造成胃内食物潴留。结果胃窦部的胃泌素细胞(G细胞)直接受到刺激,使胃泌素分泌亢进。后者与壁细胞膜上的胃泌素受体结合使壁细胞分泌胃酸,促进胃粘膜的溃疡形成。故胃溃疡患者胃窦部G细胞增多并呈高胃泌素血症(postprandial hypergastrinemia)。
3.其它因素溃疡病有时可见家族多发趋势,说明与遗传因素有关。在正常人,当食物由胃进入十二指肠球部使该处显酸性化环境时,可抑制胃内容的进一步排空。这种反馈性抑制胃排空机制在某些十二指肠溃疡患者可出现遗传性缺乏。因而胃内容排空受不到抑制,十二指肠的酸度不断增加而形成消化性溃疡。
近年研究发现,O型血者胃溃疡的发病率高于其它血型1.5~2倍。这是由于其胃粘膜细胞易受到细菌的损害。体外实验证明,引起溃疡的幽门螺旋菌(helicobacter pylori)易于攻击表面限定有O型血抗原的细胞,细菌与该型抗原接触以后进入细胞,引起感染和慢性炎症而并发溃疡(Thomas Boren,1993)。
急性溃疡胃和十二指肠粘膜有时发生急性缺损,如缺损仅累及粘膜层者称糜烂(erosion),累及粘膜下层以下时称急性溃疡(acute ulcer)。此等病变病程短而易于痊愈,并非独立的溃疡病。
急性应激性溃疡(stress ulcer)一般为表浅性溃疡,发生在胃或十二指肠粘膜,有时二处同时发生。常见于严重烧伤、创伤及败血症时,在应激反应后数小时到2周之内即可发生。其发生机制未明。可能由于上述严重损害,使机体处于应激状态,引起下丘脑功能障碍,垂体前叶分泌大量促肾上腺皮质激素,后者使胃液分泌增加,从而招致胃粘膜糜烂及溃疡形成。
三、胃癌
胃癌(carcinoma of stomach)是消化道最常见的恶性肿瘤之一。在我国不少地区的恶性肿瘤死亡统计中,胃癌居第一或第二位。胃癌好发年龄为40~60岁。男多于女,男女之比约为3:1或2:1。好发部位为胃窦部,特别是小弯侧(约占75%),胃体部则少见。
根据胃癌的病理变化进展程度分为早期胃癌与进展期(晚期)胃癌两大类。
(一)早期胃癌
癌组织浸润仅限于粘膜层及粘膜下层者均属早期胃癌(early gastric carcinoma)。所以判断早期胃癌的标准不是其面积大小而是其深度。故早期胃癌也称为粘膜内癌或表浅扩散性癌(图10-10)。早期胃癌经手术切除治疗,预后颇为良好,术后5年存活率达54.8%~72.8%。近年由于纤维胃镜活检和脱落细胞学检查方法的推广应用,早期胃癌的发现率有了明显提高(图10-11)。
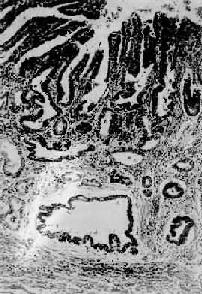
图10-10 早期胃癌
癌变局限于胃粘膜内,未超出粘膜肌层
图10-11 早期胃癌的脱落细胞
(与图10-10同一病例) 癌细胞较大,形状不一,核大,着色深
早期胃癌的肉眼形态可分为3种类型(图10-12):
1.隆起型(protruded type,Ⅰ型)肿瘤从胃粘膜表面显著隆起,有时呈息肉状。
2.表浅型(superficial type,Ⅱ型)肿瘤表面较平坦,隆起不显著。此型又可细分为:①表浅隆起型(superficial elevated type,Ⅱa型)(图10-13),②表浅平坦型(superficial flat type,Ⅱb型),③表浅凹陷型(superficial depressed type,Ⅱc型),又名癌性糜烂。
图10-12 早期胃癌各型模式图
m粘膜;mm粘膜肌;sm粘膜下;pm肌层;s浆膜
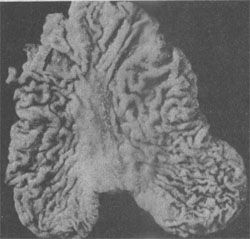
图10-13 早期胃癌(表浅隆起型Ⅱa)
在小弯侧近幽门处粘膜上有一直径约1cm大小的突出于表面的胃癌(箭头指处)
3.凹陷型(excavated type,Ⅲ型)有溃疡形成,溃疡可深达肌层。此型最为多见。
组织学分型:以管状腺癌最多见,其次为乳头状腺癌,未分化型癌最少。
(二)进展期胃癌
癌组织浸润到粘膜下层以下者均属进展期胃癌(advanced gastric carcinoma),或称之为中晚期胃癌。癌组织浸润越深,预后越差,侵至浆膜层的5年存活率较侵至肌层的明显降低。
肉眼形态可分为3型:
1.息肉型或蕈伞型(polypoid or fungating type)癌组织向粘膜表面生长,呈息肉状或蕈状,突入胃腔内(图10-14)。
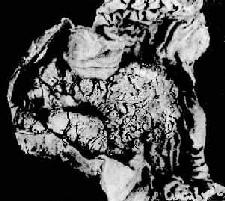
图10-14 息肉型胃癌
肿块呈息肉状突起于胃腔内
2.溃疡型(ulcerative type) 部分癌组织坏死脱落,形成溃疡。溃疡一般多呈皿状,有的边缘隆起,如火山口状(图10-15)。
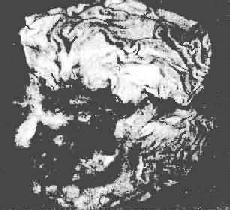
图10-15 溃疡型胃癌
肿块中央溃烂,形成边缘隆起、形状不规则的溃疡
溃疡型胃癌的溃疡与良性胃消化性溃疡大体形态的鉴别见表10-1。
表10-1 良、恶性溃疡的肉眼形态鉴别
| 良性溃疡(溃疡病) | 恶性溃疡(溃疡型胃癌) | |
| 外 形 | 圆形或椭圆形 | 不整形、皿状或火山口状 |
| 大 小 | 溃疡直径一般<2cm | 溃疡直径常>2cm |
| 深 度 | 较深 | 较浅 |
| 边 缘 | 整齐、不隆起 | 不整齐、隆起 |
| 底 部 | 较平坦 | 凹凸不平,有坏死出血 |
| 周围粘膜 | 皱襞向溃疡集中 | 皱襞中断,呈结节状肥厚* |
*因粘膜下层有癌组织生长
3.浸润型(infiltrating type)癌组织向胃壁内呈局限或弥漫浸润,与周围正常组织无明显边界。当弥漫浸润时致胃壁增厚、变硬,胃腔缩小,粘膜皱襞大部消失。典型的弥漫浸润型胃癌其胃状似皮革制成的囊袋,因而有革囊胃之称(linitis plastica)(图10-16)。
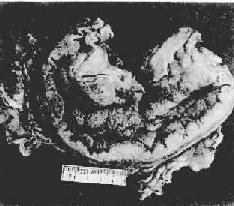
图10-16 弥漫浸润型胃癌
胃壁因癌组织的弥漫浸润而显著增厚
进展期胃癌的肉眼分型常用的还有Borrmann(B)分型。其中B1型为隆起型,B2型为限局溃疡型,B3型为浸润溃疡型,B4型为弥漫浸润型。
镜下,根据癌的组织结构,一般将进展期胃癌分为4种组织学类型:
1.腺癌(adenocarcinoma)最多见,癌细胞大多呈柱状,排列成腺腔(腺管状腺癌,glandular form)(图10-17),腺腔内出现许多乳头(乳头状腺癌,papillary form)。有的癌细胞呈立方形或圆形,由数个癌细胞形成小腺泡(腺泡状腺癌,acinar form)。此型癌组织分化较高,恶性度较低,转移较晚。
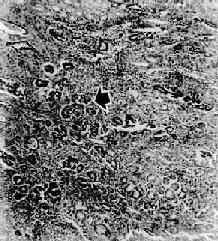
图10-17 胃腺癌
癌组织呈腺样结构,弥漫浸润于胃壁内
2.髓样癌(medullary carcinoma)癌细胞无腺样排列,细胞大而多形,异型性显著,恶性度较高,常较早地向深层浸润。
3.硬癌(scirrhous carcinoma)癌细胞较小,圆形或短梭形,呈条索状排列,多无腺管样结构,间质为大量纤维组织(图10-18)。本型恶性度较高。
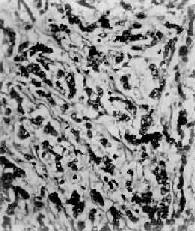
图10-18 胃硬癌
粘膜下层显著增生的纤维组织内有癌细胞浸润,癌细胞排成窄条索状
髓样癌和硬癌因癌组织多无腺腔形成,呈实体结构,故又称为实体癌或单纯癌。
4.粘液癌(mucoid carcinoma)呈腺样结构或单纯癌结构,癌细胞胞浆内出现大量偏酸性粘液,常将胞核挤压于癌细胞浆之一侧,形似戒指,故称之为印戒细胞(signet-ring cell)(图10-19)。粘液癌的恶性度高。此型因癌组织含大量粘液,肉眼上呈半透明的胶冻状故也称胶样癌(colloid carcinoma)。
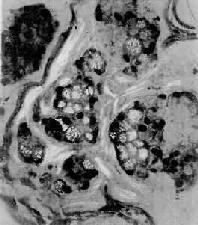
图10-19 粘液癌
癌细胞成团,胞浆内充满大量粘液,核被挤向一侧,呈印戒状
除上述分型外,也有根据胃癌细胞形态和胃癌组织中粘液性质将胃癌分为肠型和弥漫型两大类。肠型胃癌中唾液酸粘液及硫酸粘液多见,而弥漫型胃癌中中性粘液多见。肠型胃癌多见于老年患者,恶性度较低,组织学上多为乳头状腺癌或腺管状腺癌。弥漫型胃癌多见于青年人,恶性度较高,组织学上多为粘液癌及未分化癌。
需要指出,许多胃癌的组织学结构不是单一类型,在同一胃癌标本中往往有两种组织类型同时存在。发生在贲门部的胃癌,可为兼有腺上皮及鳞状上皮的腺棘皮癌(adenoacanthoma)及鳞状细胞癌。
【扩散途径】
1.直接扩散浸润到胃浆膜层的癌组织,可直接扩散至邻近器官和组织,如肝、胰腺及大网膜等。
2.淋巴道转移为胃癌转移的主要途径,首先转移到局部淋巴结,其中以胃小弯侧的胃冠状静脉旁淋巴结及幽门下淋巴结最为多见。由前者可进一步扩散到腹主动脉旁淋巴结、肝门处淋巴结而达肝内;由后者可到达胰头上方及肠系膜根部淋巴结。转移到胃大弯淋巴结的癌瘤可进一步扩散到大网膜淋巴结。晚期,癌细胞可经胸导管转移到锁骨上淋巴结,且以左锁骨上淋巴结多见。
3.血道转移多在晚期,常经门静脉转移到肝,其次可转移至肺、骨及脑。
4.种植性转移胃癌特别是胃粘液癌细胞浸润至胃浆膜后,可脱落到腹腔,种植于腹壁及盆腔器官腹膜上。有时在卵巢形成转移性粘液癌,称Krukenberg瘤。
【胃癌的组织发生】
1.胃癌的细胞来源从早期微小胃癌的形态学研究推测,胃癌主要发生自胃腺颈部的干细胞。此处腺上皮的再生修复特别活跃,可向胃上皮及肠上皮分化,癌变常由此部位开始。
2.肠上皮化生与癌变在早期胃癌标本可观察到肠上皮化生(大肠型)过渡到肠型胃癌的理象。大肠型化生在胃癌癌旁粘膜上皮的检出率常可达88.2%。有人推测癌变机制可能由于这种肠上皮化生的细胞对致癌物质的吸收增强,并且发现肠上皮化生细胞及癌细胞的胞浆中均有高活性的氨基酞酶(amino-peptidase),而正常胃粘膜中,该酶不显活性。这种变异很可能构成癌变的基础。
3.不典型增生与癌变 胃癌时重度不典型增生(severe atypical hyperplasia)多出现在癌旁,有的并与癌变呈移行关系。目前认为重度不典型增生为具有癌变潜能的一种癌前病变。
【病因】
至今未明。人类胃癌的发生有一定的地理分布特点。如在日本、中国、冰岛、智利及芬兰等国家的发病率远较美国及西欧国家为高。这可能与各国家、民族的饮食习惯及各地区的土壤地质因素有关。据调查,胃癌的发生和大量摄取鱼、肉类熏制食品有关。用黄曲霉毒素污染或含亚硝酸盐的食物饲喂动物也可诱发胃癌。在日本曾有人提出胃癌的高发与居民食用的稻米经滑石粉处理有关。因滑石粉内含有致癌作用的石棉纤维。近年,由于日本饮食习惯的改变,其胃癌的发生率有下降趋势。
【其它肿瘤】
平滑肌瘤胃肠是除子宫外的另一平滑肌瘤多发部位。其中,以胃为最多,其次是小肠。此瘤常可突入胃肠腔,也可呈巨大团块由浆膜向外突出,此时则不引起胃肠症状。肿瘤组织由交织的束状平滑肌细胞构成,细胞两端有长突起,胞核钝圆,也偶见圆形或多角形细胞。如瘤细胞是不典型或奇型的平滑肌细胞则称为平滑肌母细胞瘤。在组织学上区分平滑肌瘤及平滑肌母细胞瘤较为困难。后者虽有瘤细胞的不典型增生,但仍不能视为恶性。属于恶性的平滑肌肉瘤除显示多数瘤细胞核呈多型性并可有奇异形态的巨细胞外,较多的核分裂像也常是恶性的指征。平滑肌肉瘤常由血行转移,也可直接浸润、扩散到腹膜表面。
恶性淋巴瘤结外的恶性淋巴瘤最常发生在胃,其次为肠。胃之原发性恶性淋巴瘤在临床上甚难与胃癌区别。本瘤术后5年存活率较胃癌为高。外观,常呈扁平盘状突起,边缘清楚。有时肉眼上与癌难以区别。
四、阑尾炎
阑尾炎(appendicitis)是一种常见病。临床上常有右下腹部疼痛、体温升高、呕吐和中性粒细胞增多等表现。
【病因和发病机制】
细菌感染和阑尾腔的阻塞是阑尾炎发病的二个主要因素。阑尾是一条细长的盲管,管腔狭小,易潴留来自肠腔的粪便及细菌。阑尾壁富于神经装置(如肌神经丛等),阑尾根部并有类似括约肌的结构,故受刺激时易于收缩使管腔更为狭窄。阑尾动脉为回结肠动脉的终末分支,是一条终动脉,故因刺激发生挛缩或有阻塞时,常招致阑尾的缺血甚至坏死。
阑尾炎因细菌感染引起,但无特定的病原菌。通常在阑尾腔内能找到大肠杆菌、肠球菌及链球菌等,但必须在阑尾粘膜发生损害之后,这些细菌才能侵入引起阑尾炎。阑尾腔可因粪石、寄生虫等造成机械性阻塞,也可因各种刺激引起阑尾痉挛,引起阑尾壁的血液循环障碍造成粘膜损害,有利于细菌感染而引起阑尾炎。
【病变】
1.急性阑尾炎,有3种主要类型
(1)急性单纯性阑尾炎(acute simple appendicitis):为早期的阑尾炎,病变多只限于阑尾粘膜或粘膜下层。肉眼观,阑尾轻度肿胀、浆膜面充血、失去正常光泽。镜下,粘膜上皮可见一个或多个缺损,并有中性粒细胞浸润和纤维素渗出(图10-20)。粘膜下各层则有炎性水肿。
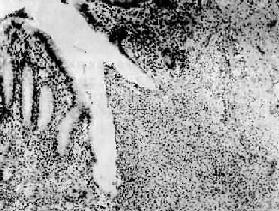
图10-20 急性单纯性阑尾炎
阑尾隐窝处粘膜上皮坏死脱落,该处有大量中性白细胞浸润
(2)急性蜂窝织炎性阑尾炎(acute phlegmonous appendicitis):或称急性化脓性阑尾炎,常由单纯性阑尾炎发展而来。肉眼观,阑尾显著肿胀,浆膜高度充血,表面覆以纤维素性渗出物。镜下,可见炎性病变呈扇面形由表浅层向深层扩延,直达肌层及浆膜层(图10-21)。阑尾壁各层皆为大量中性粒细胞弥漫浸润,并有炎性水肿及纤维素渗出。阑尾浆膜面为渗出的纤维素和中性粒细胞组成的薄膜所覆盖,即有阑尾周围炎及局限性腹膜炎表现。
图10-21 急性蜂窝织炎性
阑尾炎的发展过程模式图 图中点状区示炎性细胞浸润
(3)急性坏疽性阑尾炎(acute gangrenous appendicitis):是一种重型的阑尾炎。阑尾因内腔阻塞、积脓、腔内压力增高及阑尾系膜静脉受炎症波及而发生血栓性静脉炎等,均可引起阑尾壁血液循环障碍,以至阑尾壁发生坏死。此时,阑尾呈暗红色或黑色,常导致穿孔,引起弥漫性腹膜炎或阑尾周围脓肿。
【结局及合并症】
急性阑尾炎经过外科治疗,预后良好。只有少数病例因治疗不及时或机体抵抗力过低,出现合并症或转变为慢性阑尾炎。
合并症中主要有因阑尾穿孔引起的急性弥漫性腹膜炎和阑尾周围脓肿。有时因并发阑尾系膜静脉的血栓静脉炎,细菌或脱落的含菌血栓可循门静脉血流入肝而形成转移性肝脓肿。如果阑尾近端发生阻塞,远端常高度膨胀,形成囊肿。其内容物可为脓汁(阑尾积脓)或为粘液(阑尾粘液囊肿,mucocele)。粘液囊肿破裂,粘液进入腹腔,可在腹膜上形成假粘液瘤(pseudomyxoma)。
2.慢性阑尾炎多为急性阑尾炎转变而来,也可开始即呈慢性经过。主要病变为阑尾壁的不同程度纤维化及慢性炎细胞浸润等。临床上时有右下腹疼痛。
五、非特异性肠炎
非特异性肠炎包括肠多种非特异性炎症性疾病,大都病因不明,在病理学上无特异性变化,故称为非特异性肠炎,主要有3种。
(一)急性出血坏死性肠炎
急性出血性坏死性肠炎(acute hemorrhagic enteritis)或简称坏死性肠炎。是以小肠急性出血坏死性炎症为主要病变的儿科急症。常发生于婴儿,临床主要表现为腹痛、便血、发热、呕吐、腹泻等,重者常引起休克致死。
【病变】
肠壁发生明显的出血及坏死,常呈节段性分布(图10-22),以空肠及回肠最为多见且严重。病变肠壁增厚,粘膜肿胀,广泛出血、坏死,表面常被覆假膜。病变粘膜与正常粘膜分界清楚,常继发溃疡形成,溃疡深者可引起肠穿孔。粘膜下层除广泛出血外,发生严重水肿及炎细胞浸润。肌层平滑肌纤维断裂并可发生坏死。
图10-22 急性出血性坏死性肠炎
空肠粘膜呈节段性出血、坏死改变
【病因和发病机制】
至今不清。有较多的报告提出,本病是一非特异性感染如细菌、病毒或其分解产物所引起的激烈的变态反应(Schwartzman反应)性疾病。肠组织的出血、水肿、坏死及炎症反应均符合该种变态反应的表现。此外,一些学者在本病患者肠腔中发现一种可产生剧烈毒素的F型厌气菌,其B毒素有引起强烈的溶血、坏死作用。但此种细菌的病因作用尚待进一步证实。
(二)局限性肠炎
局限性肠炎(regional enteritis)亦称Crohn病,是一种病因未明的肠疾病。病变主要累及回肠末端,其次为结肠、近端回肠和空肠等处。消化道其他处偶见发生。因病变局限且呈节段性分布,故称为局限性肠炎。
临床表现主要为腹痛、腹泻、腹部肿块、肠瘘形成及肠梗阻等症状。本病呈慢性经过,病程较长。经治疗后病情可缓解,但常复发,不见癌变。本病与肠结核、慢性溃疡性结肠炎等常甚难鉴别。
【病变】
肉眼观,病变处肠壁变厚、变硬,肠粘膜高度水肿而呈块状增厚,如鹅卵石状或息肉状(图10-23)。粘膜面有裂隙及匐行溃疡,裂隙狭长而深入呈穿通性,重者可引起慢性肠穿孔及瘘管形成。穿孔后常形成腹腔积脓。肠壁增厚常致肠腔狭窄,引起慢性肠梗阻。病变肠管易与邻近肠管或腹壁粘连。肠壁粘合成团,颇似回盲部增殖型结核。
图10-23 结肠局限性肠炎
肠壁增厚、水肿,肠粘膜水肿,皱襞增宽、增厚,有处呈息肉状(箭头所指)
图10-24 局限性肠炎
肠粘膜下层明显增厚,淋巴管高度扩张和多量单核细胞及淋巴细胞浸润
镜下,本病的病变复杂多样,裂隙状溃疡表面覆以坏死组织,其下肠壁各层组织中可见大量淋巴细胞、单核细胞及中性粒细胞浸润。肠粘膜下层增厚、水肿,其中有多数扩张的淋巴管。有的部位粘膜下淋巴组织增生并有淋巴滤泡形成。部分病例在肠壁内见有由类上皮细胞、多核巨细胞形成的肉芽肿。肉芽肿中心不发生干酪样坏死。据此可与结核性肉芽肿鉴别。
【病因和发病机制】
迄今不明。近年发现本病伴有免疫异常现象。在患者的血液中可测到抗结肠抗体。在病变部位用免疫荧光和酶标方法证明有这种免疫复合物存在(抗原存在于患者肠上皮细胞),并有补体C3的沉积,表明可能因发生似Arthus反应而损伤肠组织,造成本病。关于肠壁水肿、溃疡的形成多认为系肠淋巴回流障碍进而导致血液循环障碍所致。
(三)慢性溃疡性结肠炎
慢性溃疡性结肠炎(chronic ulcerative colitis)是结肠的一种慢性炎症。因其病因不明,故又有特发性溃疡性结肠炎(idiopathic ulcerative colitis)之称。病变可累及结肠各段,偶见于回肠。本病病变部位多以直肠、乙状结肠为主。这可能是由于该处镜检诊断较为方便之故。临床上有腹痛、腹泻、便血等症状,病程有时缓解,可持续多年。
【病变】
最初结肠粘膜充血并出现点状出血,粘膜隐窝有小脓肿形成。脓肿逐渐扩大,局部肠粘膜表层坏死脱落,形成表浅小溃疡并可累及粘膜下层。溃疡可融合扩大或相互穿通形成窦道。病变进一步发展,肠粘膜可出现大片坏死并形成大的溃疡。残存的肠粘膜充血、水肿并增生形成息肉样外观,称假息肉。假息肉细长,其蒂与体无明显区别。有时溃疡穿通肠壁引起结肠周围脓肿并继发腹膜炎。病变局部的结肠可与邻近腹腔器官发生粘连。
镜下,早期可见肠粘膜隐窝处有小脓肿形成,粘膜及粘膜下层可见中性粒细胞、淋巴细胞、浆细胞及嗜酸性粒细胞浸润。溃疡底部有时可见急性血管炎,血管壁呈纤维素样坏死。溃疡边缘假息肉形成处的肠粘膜上皮可见有不典型增生,提示有癌变的可能。晚期病变区肠壁有大量纤维组织增生。
【合并症】
本病除可引起结肠周围脓肿、腹膜炎外,尚可合并肠癌,且一般为多发性肠癌。癌变率决定于病程长短及病变范围。一般患病15年以上者癌变率达5%~10%。此外,在暴发型病例,结肠可因中毒丧失蠕动功能而发生麻痹性扩张,故有急性中毒性巨结肠之称。
【病因】
本病的病因不明,现多认为是一种自身免疫性疾病。据报导,在大约不到半数的患者血清中可查出抗自身结肠细胞抗体。这种自身抗体可与结肠组织浸液或由大肠杆菌E.coli-14提出的多糖类抗原起交叉反应。这种交叉反应结果可引起肠粘膜的免疫性损伤。但也有在正常人血清中检出此类抗体的报道。总之,造成本病结肠粘膜破坏及溃疡形成的免疫学机制目前仍不清楚。
六、菌群失调性假膜性肠炎
又名抗生素性肠炎(antibiotic associated enteritis),常由于长期使用广谱抗生素造成肠道菌群失调所致。病原可能非单一因素。过去认为抗药性金黄色葡萄球菌为主要的病原体,以后则认为病原菌是一种梭状芽胞杆菌(clostridium difficile),该菌在正常情况下存在于肠腔内,当菌群失调时则出现异常增生,其毒素可引起肠粘膜上皮细胞变性坏死。坏死的肠粘膜与渗出的纤维蛋白形成假膜而成为假膜性肠炎。肠的各段均可受累。病变特点有:肠壁充血水肿,常见出血、粘膜表面坏死和假膜形成。假膜脱落后,可形成表浅而不规则的溃疡。肠腔扩张,腔内充满液体,可致水样腹泻、脱水和休克。
除上述细菌性肠炎外也可因长期使用抗生素消除了正常肠道菌群中与霉菌相拮抗的细菌,使霉菌得以大量繁殖而引起肠霉菌病。如肠曲菌病、毛霉菌病和白色念珠菌病等。此时肠粘膜表面坏死,有溃疡形成及出血。肠白色念珠菌病时在肠粘膜表面还可形成灰白色假膜。
七、缺血性肠病
缺血性肠病(ischemic bowel disease)是因肠壁缺血、乏氧,最终发生梗死的疾病。本病多见于患动脉硬化,心功能不全的老年患者。病变多以结肠脾曲为中心呈节段性发生。造成结肠缺血的直接原因多为肠系膜动、静脉,特别是肠系膜上动脉因粥样硬化或血栓形成引起的血管闭塞及狭窄。心力衰竭、休克引起血压降低,肠局部供血不足也可成为发病原因。
病变早期肠粘膜及粘膜下层出现出血及水肿,粘膜呈暗红色。伴随病程的进展及病变的加重,表层粘膜坏死、溃疡形成。病变严重者,肠壁全层坏死(透壁性梗死transmural infarction),甚至引起肠壁破裂、腹膜炎、休克致死。梗死面积小者可不穿透肠壁,局部发生纤维化。病变自愈后可因瘢痕形成引起肠狭窄。
八、吸收不良综合征
吸收不良综合征(malabsorption syndrome)系指广义的各种营养成分通过小肠粘膜吸收发生障碍,可概分为原发性及继发性二类。
1.原发性吸收不良综合征:又称为原发性腹泻(primary sprue),属于此类者有脂肪痢(celiac sprue)及热带性腹泻(tropical sprue)二种。脂肪痢发生在幼儿者称小儿脂痢病(celiac disease),发生在成人则称为特发性脂痢(idiopathic steatorrhea)。患者消瘦,营养不良,腹泻,呈脂肪便。本病病因可能与遗传有关的代谢异常及免疫因素有关。据报道约有80%本病患者带有人白细胞抗原HLA-B8、HLA-DW3。人食谷物后其麸质中所含的麦胶蛋白肽(gliadin peptides)在肠中水解时需要有关的肽酶,如患者肠粘膜上皮缺少此酶,则未被水解的麦胶蛋白刺激肠粘膜引起本病。也有认为本病是麸质过敏性肠病(gluten sensitive enteropathy),过敏原是麦胶蛋白。将麦胶蛋白加到体外培养的空肠粘膜上,可见引起组织内T细胞增生。本病患者空肠粘膜上皮的绒毛及微绒毛呈明显萎缩,粘膜表面扁平。粘膜固有层显慢性炎性改变,有淋巴细胞、浆细胞,有时有嗜酸性粒细胞浸润。如患者饮食不含麸质类食物,则上述肠粘膜病变可以恢复。
热带性腹泻的临床表现与病理改变均与成人脂痢相同,只是病变较轻,服叶酸制剂可治愈。
2.继发性吸收不良综合征本病较原发性者多见。多见于胃肠手术后,肠的炎症及肿瘤疾患,胆道及胰腺疾病等时。患者显全身症状,出现消瘦、贫血等营养不良症状,脂肪吸收障碍明显,患者腹泻,便中出现脂肪及蛋白质。
九、Whipple病
又称肠源性脂肪代谢障碍症(intestinal lipodystrophy),多见于中年男子。患者腹泻并有脂肪便、消瘦、衰弱,有时出现游走性多发性关节炎。病理改变为小肠粘膜脂肪沉着,粘膜肥厚隆起,绒毛粗大变硬,小肠浆膜面也有脂肪沉着。镜下见粘膜下淋巴管扩张,粘膜固有层出现泡沫状巨噬细胞,胞浆内含脂质及PAS阳性物(糖蛋白)。电镜下可见细胞浆内含有镰状包涵体及杆菌样小体。PAS反应阳性。上述病变除在小肠外还见于肠系膜淋巴结及肝、脾等其他器官。由于电镜下在病变组织中查出杆菌样小体,而且用抗生素治疗收到一定效果,故近年来认为本病可能是一种传染病,但未能从患者的送检材料中培养出特殊病原体。
十、肠梗阻
由于肠内及肠外各种原因引起的小肠肠道机械性堵塞称为肠梗阻(intestinal obstruction,ileus)。
【病因及分类】
按发生原因可将肠梗阻分为机械性及功能性二大类。
机械性肠梗阻:由肠腔内的堵塞或由肠外部病变对肠的压迫、牵拉引起。肠腔内的堵塞因素常见的有蛔虫团、肿瘤、胎粪、异物及小肠先天性狭窄或闭锁。造成梗阻的肠外因素有腹膜炎后引起的肠管粘连或肠管浆膜间的纤维带形成、肠疝时疝囊内肠管的扭转或套叠,腹腔内肿瘤的压迫等。
功能性肠梗阻:因肠壁的神经肌肉麻痹引起的肠蠕动迟缓以至停滞,引起功能性肠梗阻,又称麻痹性肠梗阻(paralytic ileus)。多见于开腹手术后或急性腹膜炎时。与此相对的有小肠发生神经肌肉痉挛时,可招致肠腔闭塞,引起痉挛性肠梗阻(spastic ileus)。
【病变】
急性肠梗阻时在梗阻处上端小肠开始有一过性蠕动增强,以后则肠管麻痹、扩张。肠壁变薄,肠腔内含大量粪液,适于细菌繁殖。梗阻如位于小肠上段,常引起剧烈呕吐,有严重水及电解质丧失。由于肠内容停滞及细菌感染,肠粘膜有炎症反应,偶亦见溃疡形成甚至发生肠穿孔。如梗阻时间较长,因肠血液循环障碍,可导致肠出血和坏死。坏死穿孔后形成弥漫性腹膜炎。
十一、肠先天性发育异常疾病
(一)先天性巨结肠
先天性巨结肠(congenital megacolon)又称Hirschsprung病,是指结肠的一部分发生显着扩张与肥大。多见于1~3岁男性婴幼儿,被认为是一种家族性疾病,但传递的准确因素尚未明确。据报导约有2%的Down综合征患儿伴有本病。本病有时也合并其它先天性异常病变。患儿于出生后不久至数月内即出现便秘症状,严重者腹部膨隆,有呼吸困难及下肢静脉曲张等症状。
【病变】
结肠的一部或全部显着扩张、肥厚,结肠袋消失。扩大肠段可比正常增大数倍,与未扩大肠段分界明显。扩张肥厚的肠管内积有大量气体、粪便或粪石。由于干涸粪便及粪石长期压迫侵蚀肠粘膜,引起粘膜溃疡、炎症甚至坏死。与扩张肠段邻接的远端肠管显得细小,常称之为狭窄段。该处肠粘膜下神经丛及肌间神经丛中的神经节细胞显著减少或缺如。由此可引起乙酰胆碱及其酶类在局部积存,可用组织化学方法显示。
【病因和发病机制】
本病是一种先天性疾病。在乙状结肠下段或直肠上段肠壁肌间丛神经节细胞先天性缺如(aganglionosis)或减少,致使来自骶髓的副交感神经节前纤维不能与神经节细胞形成突触并发出节后纤维,而直接由节前纤维进入肠壁。这种没有神经节细胞调节的副交感神经节前纤维引起了肠壁的收缩与痉挛(狭窄段)。结果招致上段肠管的粪便停滞,表现顽固的便秘,最终该段肠管发生肥大与扩张(扩张段)(图10-25)。因此手术切除缺乏神经节细胞的狭窄段肠管可望治愈本病。
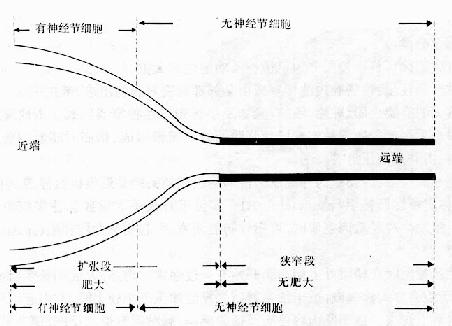
图10-25先天性巨结肠的发生示意图 (仿Budian M)
(二)肠憩室
肠憩室多为先天性憩室(congenital diverticulun)。在成人位于回盲瓣上方约1m处,在新生儿则位于0.3~0.5m处的迴肠部分,常发生在肠系膜附着部的对侧,呈指状膨出(图10-26)。憩室长度为0.3~12cm。此种憩室来自胚胎期的脐肠管(ductus omphaloentericus),正常时该管在胚胎第8周即行退化消失,但此时则不退化,残留而成憩室。如憩室发生在脐肠管的肠侧称为Meckel憩室(Meckel diverticulum),发生在脐侧称为脐瘘(umbilical fistula)。如该管全段残留未闭则称为脐肠瘘(fistula omphaloenterica),如两端闭锁中段未闭形成囊泡时则为肠管囊泡(enterocyst)。

图10-26 小肠憩室
憩室内壁多有与迴肠同样的粘膜,有时为胃或大肠粘膜。如系胃粘膜覆盖时,相近的肠粘膜易发生消化性溃疡。由于憩室内潴留的肠内容物发酵或刺激可引起该处炎症,称憩室炎(diverticulitis),症状类似阑尾炎。
后天性肠憩室较为少见,多在肠内压亢进,肠系膜脂肪组织减少等时发生。其部位与先天性者相反,常位于肠管的肠系膜附着部。
十二、肠肿瘤
(一)大肠息肉
突出于粘膜面、呈球形或卵圆形根部有蒂的肿块称为息肉(polyp)。大肠息肉是常见的一类良性肿物,其中有的属于粘膜的增生性改变(如增生性息肉),有的属于腺瘤(如腺瘤性息肉,乳头状息肉)。后一类与大肠癌的关系密切,有重要意义。
1.腺瘤性息肉(adenomatous polyp) 此种息肉从组织学结构实系腺瘤,故又称息肉状腺瘤(polypoid adenoma),较为常见。其中大约有75%发生在直肠及乙状结肠。多为单个,少数为多发。息肉直径一般在2cm以内,大多有蒂,状如草莓,色红,易出血。广基无蒂者体积较大。组织学上可见由增生的肠粘膜腺体组成。上皮细胞一般分化良好,偶见细胞有异型性。但增生的腺上皮细胞并不侵入粘膜肌层。此型发生癌变者并不多见。
2.乳头状腺瘤(papillary adenoma) 亦称绒毛状腺瘤(villous adenoma),少见,常单发,表面呈乳头状或绒毛状。基底部宽,无蒂或有极短的蒂。体积一般比腺瘤样息肉大。乳头状或绒毛状突起的表面由一层或多层柱状上皮被覆,可有不同程度的异型性。此型腺瘤的癌变率高于腺瘤样息肉。
有些腺瘤中,可见乳头状腺瘤与腺瘤样息肉的结构并存,称之为混合型腺瘤。此型的生物学行为与乳头状腺瘤近似。
3.结胞多发性息肉病(polyposis coli) 呈家族性发生,故又称家族性息肉病(familial polyposis),为一种显性遗传病。常好发于青年。肉眼可见在大肠粘膜上有许多散在的约黄豆粒大的小息肉群生(图10-27)。息肉一般无蒂,数目可多达数十或更多。镜下结构与腺瘤样息肉相同。临床症状均为腹痛、腹泻及便血。家族性息肉病易于癌变,据报导常在出现息肉症状大约15年后发生大肠癌。
图10-27 结肠多发性腺瘤性息肉
肠粘膜面见多数绿豆至黄豆粒大的小息肉
上述三种息肉均为腺瘤性质,此外还有一些非瘤性息肉,主要者如下。
增生性息肉(hyperplastic polyp):较为常见,体积较小,属粘膜增生性改变。增生的腺体规整,有旺盛的粘液分泌但无瘤样改变,可自行消失。
炎性息肉(inflammatory polyp);较为少见,一般常可以自行消失,不发生癌变。属于此类者如幼年性息肉(juvenile polyp),多见于儿童。息肉为单发性平滑圆形结节,组织学结构为肠粘膜上皮增生,有丰富的血管纤维间质及炎性细胞浸润。此类息肉也常见于血吸虫病患者。
Peutz-Jeghers综合征:为一种显性遗传疾病。在口唇及口颊粘膜等处有黑色素沉着,同时在肠发生多发性息肉。息肉具有错构瘤性质。发展成肠癌者甚为罕见。临床上主要有便血,有时有肠套叠症状。
(二)类癌
消化道类癌(carcinoid)可发生在阑尾、小肠、直肠、胃、十二指肠等处。其中发生在阑尾者最为多见,其次为小肠。由于癌细胞含有神经分泌颗粒,其中有氨基酸脱羧酶,可消耗胺前体物质,故属于APUD系统,本肿瘤亦为APUDoma之一种。
【病变】
本瘤多发生自肠粘膜腺体的Kultschitsky细胞。这种细胞起源于神经外胚层,呈嗜铬性(经重铬酸钾处理染成明显黄色)。它还有强烈的嗜银性和(或)亲银性。用硝酸银液染色可见胞浆内出现棕黑色银反应颗粒。所以,本肿瘤又称为嗜银瘤。肉眼上,瘤体不大,直径很少超过2cm,色淡黄,切面呈均质状。在小肠常为多发性,有时环绕阑尾及肠壁。
镜下见瘤细胞较小,形态一致,为圆形或多角形,核小而规则。胞浆轻度嗜碱性,有嗜银颗粒,颗粒内含5-羟色胺。瘤细胞形成实体状或索状胞巢,有时可见其中有假菊形团样结构(图10-28)。电镜观察可见瘤细胞浆内有直径100~300nm的球形分泌颗粒,颗粒中有有一电子致密核心,颗粒周边有高电子密度的界膜。瘤常限于粘膜及粘膜下层,少数可达浆膜层。发生在阑尾的类癌一般恶性度较低,转移率为3%。但发生在其他肠段及其他部位的类癌,则有约2/3为恶性者,并常转移到肝。
图10-28 结肠类癌
根据类癌细胞胞浆颗粒的银染色反应可将本瘤分为嗜银性(argyrophil)及亲银性(argentaffine)二种。后者亦称为银还原性,此时以氨银液处理肿瘤细胞,胞浆内可见有银颗粒沉着。嗜银性者还需在氨银液内加还原剂作用后在瘤细胞浆内出现银颗粒。亲银性阳性的瘤细胞,嗜银性亦为阳性。Williams及Sandler等根据银染色反应及类癌发生部位将其分为:
前肠型:发生在胃、十二指肠、胰腺、及支气管、嗜银反应+,亲银反应-。
中肠型:发生在小肠,横结肠及升结肠,亲银反应+。
后肠型:发生在降结肠及直肠,亲银反应-,嗜银反应±。
类癌综合征多见于类癌肝转移患者。出现颜面潮红,腹泻,哮喘发作,四肢抽搐,厥冷,休克状态以及心瓣膜功能障碍。产生上述综合征的主要原因是类癌细胞分泌多量5-羟色胺、组胺、缓激肽等生物活性物质作用于血管壁、胃肠、支气管平滑肌及心瓣膜所致。患者尿中可检出5-羟色胺降解产物5-羟吲哚醋酸。
(三)十二指肠壶腹癌
亦称vater壶腹癌。壶腹部的粘膜上皮包括该处的总胆管、胰管和十二指肠粘膜及粘膜下的腺体等上皮。上述各处均可成为癌的来源,故确切地应称之为壶腹部周围癌。本癌在形态结构上可为腺癌、乳头状腺癌及少见的单纯癌、粘液癌等。患者常因肿瘤阻塞总胆管而出现黄疸。
(四)大肠癌
大肠癌(carcinoma of large intestine)在我国的发病率与国外发达国家相比还处于较低水平。但近年由于饮食结构变化,本癌的发病有增加趋势,在消化道的癌中仅次于胃癌。此癌如能早期发现并及时治疗,其术后5年存活率可达90%,可见早期发现的重要性。患者多为老年人,但中青年人发生率在逐渐上升。患者常有贫血、消瘦,大便次数增多、变形,并有粘液血便。有时出现腹部肿块与肠梗阻症状。梗阻常见于降结肠癌及直肠癌,盲肠及升结肠癌往往不发生梗阻症状。这可能与发生在大肠左、右侧的癌的病理形态不同有关。
【病变】
发生部位:大肠癌的好发部位以直肠为最多,其次为乙状结肠,两者可占全部病例的2/3以上。其次是盲肠、升结肠、降结肠、横结肠。据调查大肠癌在近肛侧的大肠远端之所以增多可能与临床就用直肠乙状结肠镜检查有关。少数病例为多中心性生长,此常由多发性息肉癌变而来。
肉眼观:一般可分为4型。
1.隆起型 肿瘤向肠腔内突出,又可分为隆起息肉型(图10-29)及盘状型两个亚型。镜下多为分化成熟的腺癌(图10-30)。

图10-29 直肠癌(隆起息肉型)
图10-30 直肠腺癌
癌细胞分化较好,癌巢大多呈腺样结构(Ⅱ73-3057)
2.溃疡型肿瘤表面形成明显的较深溃疡。根据溃疡外形及生长情况又可分为局限溃疡型及浸润溃疡型2个亚型。局限溃疡型肿瘤外观似火山口状,中央坏死,有不规则形深溃疡形成。溃疡边缘呈围堤状隆起于粘膜面,边界多尚清楚。浸润溃疡型溃疡底大,向肠壁深部浸润生长,与周围正常组织分界不清。溃疡边缘为斜坡状隆起的肠粘膜。
3.浸润型肿瘤向肠壁深层弥漫浸润,常累及肠管全周,使局部肠壁增厚,表面常无明显溃疡。有时肿瘤伴纤维组织增生,可使肠管管腔周径缩小,形成环状狭窄,亦称环状型。
4.胶样型肿瘤外观及切面均呈半透明胶冻状。镜下为粘液腺癌或弥漫浸润的印戒细胞癌。此型较少见,主要发生于直肠,多见于青年人,预后较差。
大肠癌的肉眼形态在左、右侧大肠有明显的不同。左侧多为浸润型,引起肠壁环形狭窄,早期出现梗阻症状。右侧多为隆起息肉型,沿盲肠及升结肠的一侧壁生长、蔓延。一般无梗阻症状。
【组织学类型】
1.乳头状腺癌癌细胞为柱状上皮,排列成细乳头状,乳头内间质很少。多为高分化型。
2.管状腺癌癌细胞排列成腺管状。根据其分化程度可分为:高分化、中等分化及低分化三级。
3.粘液腺癌常有二种类型。一种表现为大片粘液湖形成,其中漂浮小堆癌细胞;另一种表现为囊腺状结构,囊内充满粘液,囊壁衬以分化较好的粘液柱状上皮。此类粘液腺癌常伴有高分化腺癌或乳头状腺癌。
4.印戒细胞癌肿瘤由弥漫成片的印戒细胞构成,不形成腺管状结构。有时可伴有少量细胞外粘液。
5.未分化癌癌细胞常较小,形态较一致,细胞弥漫成片或成团。无腺上皮分化,不形成腺管状结构。
6.腺鳞癌 肿瘤组织具有腺癌及鳞癌二种结构。
7.鳞状细胞癌多发生在直肠肛门附近的被覆鳞状上皮。为数较少。
大肠癌组织可产生一种糖蛋白,作为抗原引起患者的免疫反应。此种抗原称为癌胚抗原(carcino-embryonic antigen,CEA)。现知CEA可广泛存在于内胚叶起源的消化系统癌中(胃、小肠、大肠、肝、胰等处的癌),也存在于正常胚胎的消化管组织中。在正常人血清中也可有微量存在。因此,血清中检出CEA并不能作为确诊大肠癌的根据。但测定CEA有助于观察患者癌肿的消长,例如切除肿瘤后CEA水平下降,以后CEA再度上升则提示癌瘤复发或转移。
【分期】
Dukes 曾根据大肠癌病变在肠壁的扩散范围以及是否转移到局部淋巴结将大肠癌分为三期(图10-31)。
(图缺)
图10-31 Dukes肠癌分期示意图
A癌生长限于肠壁,无淋巴结转移
B癌直接扩延至肠周组织,无淋巴结转移
C?局部淋巴结转移
A期:癌组织直接侵入粘膜下层或肌层,但未穿透肌层,也未累及淋巴结。
B期:癌组织已超过肌层,扩延到肠周组织,但仍未累及淋巴结。
C期:除有上述改变外,癌已发生淋巴结转移。
A期经手术可以治愈。B期术后5年存活率为70%,C期降至30%。上述Dukes分类的缺陷是未反映早期大肠癌及有远隔器官转移的晚期大肠癌。故近年又有将A期进一步划分,A1期为早期大肠癌。在C期之后增加了D期,此期有远隔器官的癌转移。
【扩散蔓延】
1.局部扩散分化成熟的大肠癌生长缓慢,肠壁的环肌及纵肌又可限制其扩散。故在癌组织侵入肌层前,极少有淋巴结及静脉的受累。当癌已浸润到浆膜后,可直接蔓延到邻近器官,如前列腺、膀胱、腹膜及腹后壁。
2.淋巴道转移早期肿瘤可沿肠壁神经周围的淋巴间隙扩散,由淋巴管转移到淋巴结。结肠淋巴结可分为结肠上、旁、中间和终末淋巴结4组,结肠癌时均可有转移。直肠癌首先转移到直肠旁淋巴结,以后再扩散,侵入盆腔和肛周组织。
3.血道转移晚期大肠癌可经血行转移到肝、肺、骨等处。肝转移时,转移癌的部位与原发部位有关。一般右侧结胞癌多转移到肝右叶,左侧结肠癌则左、右肝叶均可转移。
【病因】
关于大肠癌的病因,在环境及遗传二种因素值得注意。在遗传因素上,曾有大肠癌家族性高发的报告。另外,在遗传性多发性大肠息肉病的患者鉴定出一种单基因突变体,据称后者与息肉的癌变有关。近年研究报道,位于第17染色体的称为p53的基因及位于第2染色体末端的新基因MSH2均为抗癌基因(antioncogene),在大肠癌时这种抗癌基因发生点突变而失活。结果失去了对细胞增殖的正常控制,从而发生癌变。在结肠息肉无癌变时则不见p53基因的失活(Bodmer & Vogelstein)。关于环境因素,现认为多与饮食有关。高营养低纤维质而少消化残渣的饮食与本病的发生有关。这类食物不利于有规律的排便,因此延长了肠粘膜与这类食物中可能含有的致癌物质的接触时间。流行病学调查发现,高脂肪饮食的人群有较高的大肠癌发病率。其粪便内胆汁酸、中性类固醇物质的含量以及厌氧菌菌丛比例都比低脂肪饮食的人群为高。肠道内的厌氧菌靠其含有的β-葡萄糖醛酸酶、7α-脱羟酶及胆固醇脱氢酶等把肠道内的胆汁酸、中性类固醇物质转化为促癌物质和(或)致癌物质,如形成多环芳香化合物等。β-葡萄糖醛酸苷酶又可把经肝解毒,并以葡萄糖醛酸苷形式随胆汁排至肠道的结合形式的致癌物质,重新游离出来发挥作用。
第三节 肝胆疾病
一、循环障碍
(一)门静脉阻塞
较为少见。多由于肝、胰疾病如肝硬变、肝癌、胰腺癌等压迫、侵袭肝内门静脉,以及化脓性腹膜炎,新生儿脐带感染化脓等引起门静脉的血栓形成或栓塞。门静脉完全而广泛的阻塞甚少见。其肝内分支的一支或多支阻塞可引起梗死(Zahn梗死)。
Zahn梗死又称萎缩性红色梗死,为肝内少见的循环障碍性病变。病变以局部肝淤血为主,而不是真性梗死。病变区呈圆形或长方形,暗红色,境界清楚(图10-32)。镜下为肝小叶中央区的高度淤血并有出血。局部肝细胞萎缩、坏死或消失。本病变对机体无大影响,偶可成为腹腔内出血的来源。病变恢复期可见阻塞的门静脉周围出现新吻合支。
图10-32 肠梗死
梗死区略呈长方形,色深,被膜下肝组织未发生梗死
(二)肝静脉阻塞
肝静脉阻塞一般分为二类。一类为肝静脉干至下腔静脉的阻塞,称Budd-Chiari综合征;另一类为肝内肝静脉小分支阻塞,称肝小静脉闭塞症(veno-occlusive disease)。
Budd-Chiari综合征是指肝静脉干和(或)下腔静脉的肝静脉入口处有一段完全或不完全阻塞而引起的征候群。本综合征的病因有原发及继发二种。原发性者主要是先天性血管异常,如下腔静脉膜性阻塞所致的肝静脉阻塞。继发性者可由血液凝固性积升高疾病(如红细胞增多症),肝癌及腹腔肿瘤,腹部创伤及某些口服避孕药等引起的该段静脉血栓形成所致。病理变化主要为肝淤血,肝细胞萎缩、变性以至坏死。此外,还有肝出血,即淤于肝窦内的红细胞进入窦外压力较低的Disse腔及萎缩的肝板内。慢性病例则发展为淤血性肝硬变。
二、代谢性疾病
(一)含铁血黄素沉积症
肝含铁血黄素沉积症(hemosiderosis)是指肝组织内有可染性铁的血色素沉着。含铁血黄素沉积的病因,主要是由于大量红细胞破坏,血红蛋白分解所引起,如引起溶血及肝内出血的疾病(慢性溶血性贫血)。含铁血黄素主要沉积于肝细胞内,Kupffer细胞内亦常有该色素沉着但一般较肝细胞轻。因输血引起者Kupffer细胞色素沉着则较明显。
血色病(hemchromatosis)是一种先天性铁代谢异常的全身性疾病。发病机制不明。肝病变为全身病变的一部分,表现为肝内重度含铁血黄素沉积,全肝呈铁锈色。后期伴有肝纤维化或肝硬变。
(二)糖原沉积症
糖原沉积症(glycogenosis)为先天性常染色体隐性遗传所引起的组织内糖原质的异常和量的增多,而引起沉积。主要累及肝、心、肾及肌组织,有低血糖、酮尿及发育迟缓等表现。按糖代谢过程中不同环节、不同酶的异常,现已将本症分为O~Ⅺ型,如包括亚型则更多(Schiff,1987)。
肉眼可见肝肿大,有的可达正常肝的3倍以上。颜色变淡。镜下,肝细胞明显肿大,泡浆淡染,呈疏松的颗粒状并有空亮区。冷冻切片,PAS染色可见肝细胞内红色的糖原颗粒。对淀粉酶的消化反应稳定。后期,多种类型可伴有肝纤维化或肝硬变。需要指出,确诊糖原沉积症及分型,不能单凭病理组织学改变,必须结合临床及用肝穿取新鲜标本作酶类分析。
(三)类脂质沉积症
类脂质沉积症(lipoidosis)是先天缺陷性脂质代谢障碍所致的组织内类脂质增多并沉积。主要有糖脂、磷脂及胆固醇等沉积。其发生机制,大都是由于作用于脂质分解代谢某些环节上的酶类的遗传性缺失,使其相应的底物(脂质)分解代谢不能进行而沉积在组织内。
1.糖脂沉积症糖脂 是指不含磷酸的脑苷脂及神经苷脂等脂类。它们的分解代谢障碍可分别引起脑苷脂沉积症(如高雪病)和神经节苷脂沉积症。
高雪(Gaucher)病,也称脑苷脂沉积症,是由于常染色体隐性遗传所致体内β-葡萄糖苷酶缺乏而引起的脑苷脂分解代谢障碍。主要累及肝、脾、淋巴结及骨髓等单核吞噬细胞系统。常发生在婴儿,为致命性疾病。主要病变为肝、脾肿大,脾大尤为明显,可达正常脾重的20倍。镜下,肝内聚集大量高度胀大的载脂巨噬细胞,有的胞浆呈泡沫状,有的胞浆出现红染条纹,后者排列成皱纹纸样外观,胞核小,圆形或椭圆形居于细胞中央,称为高雪细胞(图10-33)。这些细胞主要分布于小叶中央静脉附近的肝窦内和汇管区。偶见发生肝纤维化和肝硬化。
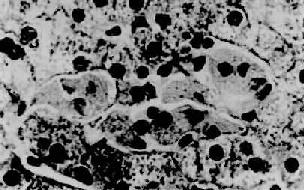
图10-33 高雪病之肝
图中央区为数个高雪细胞,位于肝窦内胞浆呈皱纹纸状外观
2.磷脂沉积症 主要为不含甘油成分的神经磷脂的增多、蓄积,又称尼曼-皮克病。
尼曼-皮克(Niemann-Pick)病又称神经磷脂沉积症。系由于常染色体隐性遗传所致的神经磷脂酶缺乏,使神经磷脂不能被水解而沉积于组织内所致。另外还可伴有其它脂质贮积。本病主要累及肝、脾、骨髓及淋巴结等器官,在儿童也侵犯神经系统。肝病变肉眼观,肝肿大。镜下,在肝窦内和汇管区有大量Kupffer细胞和巨噬细胞聚集,细胞体积肿大,胞浆呈泡沫状,核小居中,称为Pick细胞。肝细胞内也可见有脂肪,主要为中性脂肪及胆固醇。电镜下见Pick细胞内充满多数年轮样层状排列的球形包涵体。本病常发生于幼儿,预后不佳。
(四)铜代谢障碍疾病—肝豆状核变性
肝豆状核变性(hepatolenticular degeneration)又称威尔逊病(Wilson’s disease)。本病为位于第13染色体的隐性基因传递的遗传性疾病,家族性多发。患者多为儿童及青少年。本病的特点是铜代谢障碍,不能正常排出而蓄积于各器官。首先累及肝,待肝饱和后再沉积于中枢神经系统,故出现神经症状。铜也可蓄积于角膜,在角膜周围出现绿褐色环(Kayser-Fleischer环)。肝病变:在肝细胞中可见有脂褐素、铜结合蛋白、铁等沉着。铜或铜结合蛋白(如rhodamine,rubeanic acid等)可由组织化学染色检出。早期见肝细胞线粒体基质中有大颗粒或晶体沉着。肝并有急、慢性肝炎及肝硬变等病变伴发。中枢神经系统可见被壳及苍白球变性,尾状核萎缩,大脑皮质及小脑变性等。
三、病毒性肝炎
病毒性肝炎(viral hepatitis)是由肝炎病毒引起的以肝实质细胞变性坏死为主要病变的传染病。现已知肝炎有甲型、乙型、丙型、丁型及戊型5种,由各该型病毒引起。1974年以来提出的非甲非乙型肝炎(NANB型),经近年研究证明,其中大部分为丙型肝炎并检出了丙肝病毒(HCV)及其抗体,另一小部分则为戊型肝炎。肝炎在世界各地均有发病和流行,且发病率有不断升高趋势。其发病无性别差异,各种年龄均可罹患。
【病因】
从1970年起,经过20年的研究,目前对肝炎病毒已比较清楚,由最初仅知的甲型肝炎病毒和乙型肝炎病毒二种,增加到由甲到戊5种病毒(HAV~HEV)。其特点见表10-2。
表10-2 各型肝炎病毒特点
| 肝炎及其病毒分型 | 病毒类型 | 传染方式 | 克隆化年代 |
| 甲型肝炎HAV | EnterovirusRNA型 | 肠道 | 1983年(1973检出) |
| 乙型肝炎HBV | HepadnavirusDNA型 | 非肠道 | 1979年(1963检出) |
| 丙型肝炎HCV | FlavivirusRNA型 | 非肠道 | 1989年 |
| 丁型肝炎HDV | ViroidrelatedRNA型 | 非肠道 | 1986年 |
| 戊型肝炎HEV | CalcivirusRNA型 | 肠道 | 1990年 |
(采自Seeff)
在上述各型肝炎病毒中,以HBV发现最早,研究得最多。现知该病毒是由核心及外壳两部分构成的病毒颗粒(Dane颗粒)。病毒基因组DNA在肝细胞核内进行复制、转录,合成核心颗粒后,被转运到肝细胞浆内,在通过内质网和细胞膜时合成其外壳部分,并以“发芽”过程释出肝细胞。Dane颗粒的核心部分含核心抗原(HbcAg),外壳部分含表面抗原(HBsAg)。目前认为引起肝细胞免疫损害的只是HBsAg诱发的免疫反应,其中以细胞免疫反应起主要作用。HAV是一种微小的RNA病毒,在肝细胞浆内增殖,现已被列入肠道病毒,该病毒颗粒可在粪便中检出。HCV的整组基因近年也可克隆化并标记其病毒抗体。HDV是一种微小缺陷性RNA病毒,现知此种病毒为球形以HBsAg作为其外壳,只能在HBsAg阳性机体内生长,故丁型肝炎常与乙型肝炎合并存在。HEV的颗粒为球形无包膜,表面有尖钉状突起。我国学者已将HEV分离培养并建立了细胞系(Huang RT等,1992)。
【传染途径】
各型肝炎病毒均可存在于肝组织、血液、粪、尿及各种体液内。其传染方式主要是经口、经血及体液传播,但各型肝炎的传染途径各异。甲型、戊型多经口感染。常来源于饮水及食物的污染,有时呈流行性暴发。乙型、丙型经血感染,主要通过输血、输液也可通过经皮及性接触传播。丁型亦为非经口感染,常与乙型肝炎传播伴行。各型肝炎的潜伏期也不相同,如甲型15~50天,乙型60~180天。戊型2~9周。丙型7~8周。一般认为肝炎痊愈后均可获得免疫力但均不稳固(甲型者稍好),有少部分患者还可发生再感染。
【基本病变】
各型肝炎病变基本相同,都是以肝细胞的变性、坏死为主,同时伴有不同程度的炎性细胞浸润、肝细胞再生和纤维组织增生。
1.肝细胞变性、坏死
(1)胞浆疏松化和气球样变:为常见的变性病变,是由于肝细胞受损后细胞水分增多造成。开始时肝细胞肿大,胞浆疏松呈网状、半透明,称胞浆疏松化。进一步发展,肝细胞更形胀大呈球形,胞浆几完全透明,称为气球样变(ballooning degeneration)(图10-34)。电镜下,可见内质网扩张、囊泡变、核蛋白颗粒脱失;线粒体肿胀、嵴消失等。
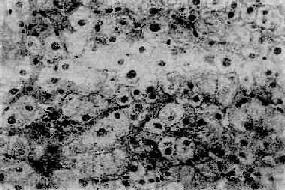
图10-34 急性病毒性肝炎
肝细胞胞浆疏松化和气球样变,肝窦受压变窄
(2)嗜酸性变及嗜酸性坏死:嗜酸性变多累及单个或几个肝细胞,散在于小叶内。肝细胞胞浆水分脱失浓缩,嗜酸性染色增强,胞浆颗粒性消失。如进一步发展,胞浆更加浓缩之外,胞核也浓缩以至消失。最后剩下深红色均一浓染的圆形小体,即所谓嗜酸性小体(acidophilic body或Councillman body)(图10-35)。上述改变称嗜酸性坏死(acidophilic necrosis),为单个细胞坏死(细胞凋谢)。
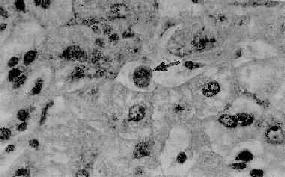
图10-35 急性病毒性肝炎
游离于肝窦内的嗜酸性小体(↑处),窦内皮细胞肿胀增生
(3)点状坏死(spotty necrosis):肝小叶内散在的灶状肝细胞坏死。每个坏死灶仅累及1至几个肝细胞。同时该处伴以炎性细胞浸润(图10-36)。
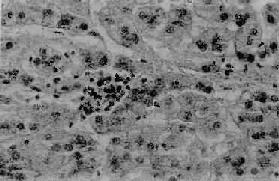
图10-36 病毒性肝炎
肝细胞点状坏死,坏死灶内有炎性细胞浸润
(4)溶解坏死(lytic necrosis)最多见,常由高度气球样变发展而来。此时胞核固缩、溶解、消失,最后细胞解体。重型肝炎时肝细胞的变性往往不明显,很快就发生此种坏死崩解。
2.炎细胞浸润肝炎时在汇管区或肝小叶内常有程度不等的炎性细胞浸润。浸润的炎细胞主要是淋巴细胞、单核细胞,有时也见少量浆细胞及中性粒细胞等。
3.间质反应性增生及肝细胞再生
(1)Kupffer细胞增生肥大:这是肝内单核吞噬细胞系统的炎性反应。增生的细胞呈梭形或多角形,胞浆丰富,突出于窦壁或自壁上脱入窦内成为游走的吞噬细胞。
(2)间叶细胞及纤维母细胞的增生:间叶细胞具有多向分化的潜能,存在于肝间质内,肝炎时可分化为组织细胞参与炎性细胞浸润。在反复发生严重坏死病例,由于大量纤维组织增生可发展成肝纤维化及肝硬变。
(3)肝细胞再生:肝细胞坏死时,邻近的肝细胞可通过直接或间接分裂而再生修复。在肝炎恢复期或慢性阶段则更为明显。再生的肝细胞体积较大,核大而染色较深,有的可有双核。慢性病例在汇管区尚可见细小胆管的增生。
上述肝炎基本病变中,肝细胞疏松化,气球样变,点状坏死及嗜酸性小体形成对于诊断普通型肝炎具有相对的特征性;而肝细胞的大片坏死、崩解则是重型肝炎的主要病变特征。
【临床病理类型】
各型肝炎病毒引起的肝炎其临床表现和病理变化基本相同。现在常用的分类是,在甲、乙、丙、丁、戊5型病毒病因分类之外,把病毒性肝炎从临床病理角度分为普通型及重型二大类。在普通型中分为急性及慢性两类。急性有急性无黄疸型及黄疸型;慢性有持续性(迁延性)及活动性。重型中又分为急性及亚急性两种。
1.急性(普通型)肝炎最常见。临床上又分为黄疸型和无黄疸型二种。我国以无黄疸型肝炎居多,其中多为乙型肝炎,一部分为丙型即过去所谓非甲非乙型的一部分。黄疸型肝炎的病变略重,病程较短,多见于甲型、丁型、戊型肝炎。两者病变基本相同,故一并叙述。
病变:广泛的肝细胞变性,以胞浆疏松化和气球样变最为普遍。坏死轻微,肝小叶内可有散在的点状坏死。嗜酸性小体的出现并非经常。由于点状坏死灶内的肝细胞索网状纤维支架保持完整而不塌陷,所以该处通过再生的肝细胞可完全恢复原来的结构和功能。汇管区及肝小叶内也有轻度的炎性细胞浸润。黄疸型者坏死灶稍多、稍重,毛细胆管管腔中有胆栓形成。
【临床病理联系】
由于肝细胞弥漫地变性肿胀,使肝体积增大,被膜紧张,为临床上肝大、肝区疼痛或压痛的原因。由于肝细胞坏死,释出细胞内的酶类入血,故血清谷丙转氨酶(SGPT)等升高,同时还可引起多种肝功能异常。肝细胞坏死较多时,胆红质的摄取、结合和分泌发生障碍,加之毛细胆管受压或有胆栓形成等则可引起黄疸。
结局:急性肝炎大多在半年内可逐渐恢复。点状坏死的肝细胞可完全再生修复。一部分病例(多为乙型、丙型肝炎)恢复较慢,需半年到一年,少数病例(约1%)可发展为慢性肝炎。极少数可恶化为重型肝炎。
2.慢性(普通型)肝炎病毒性肝炎病程持续在一年(国外定为半年)以上者即为慢性肝炎。其中乙型肝炎占绝大多数(80%),也有近年明确的丙型肝炎。按病程、肝功能情况、免疫状态及病变等的不同将慢性肝炎分为持续性(迁延性)和活动性(进展性)二种。
(1)慢性持续性肝炎(chronic persistent hepatitis,CPH):临床症状常较轻或仅有肝功能异常。镜下,肝细胞变性、坏死较急性时减轻,Kupffer细胞增生活跃,汇管区或小叶内慢性炎性细胞浸润明显。有时汇管区可因有少量结缔组织增生而变宽。肝小叶轮子廓清楚,小叶界板无破坏。肉眼观,肝体积增大,但表面平滑。此型肝炎一般发展缓慢,经过较好,大多数可以恢复,少数可转变为慢性活动性肝炎。
(2)慢性活动性肝炎(chuonic active hepatitis,CAH):此型肝炎病变较重,肝功能持续异常。镜下,肝细胞变性坏死更为广泛而严重。肝细胞坏死呈灶状或条带状,并具有以下二种特征。①小叶周边的肝细胞界板受到破坏,界板肝细胞呈灶状坏死、崩解,伴有炎性细胞浸润,称为碎片状坏死(piecemeal necrosis)(图10-37);②小叶中央静脉与汇管区之间或两个中央静脉之间出现肝细胞坏死带,称桥接坏死(bridging necrosis)。坏死区可出现肝细胞不规则再生。小叶周边部坏死区纤维组织增生呈星芒状向小叶内伸展,并与小叶内肝细胞坏死处网状纤维支架塌陷而胶原化的纤维条索相连接,形成纤维间隔而分割小叶结构。肉眼观,在肿大的肝表面,上述纤维化明显区呈不平滑颗粒状,质地较硬。此型常见于乙、丙型肝炎,除肝外,患者还有脾肿大等全身改变,如不及时治愈大都转入肝硬变。
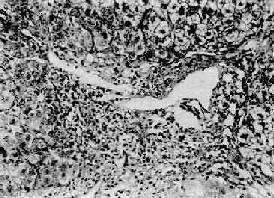
图10-37 慢性活动性肝炎
肝细胞明显气球样变和嗜酸性变,小叶界板破坏呈现碎片状坏死,门管区见炎性细胞浸润
毛玻璃样肝细胞 多见于HBsAg携带者及慢性肝炎患者的肝组织。光镜下,HE染色切片上,此等肝细胞浆内充满嗜酸性细颗粒状物质,不透明似毛玻璃样故称毛玻璃样肝细胞(图10-38)。这些细胞内含大量HBsAg,电镜下呈线状或小管状积存在内质网池内。用免疫酶标法或免疫荧光法可呈HBsAg阳性反应。(图10-39)。
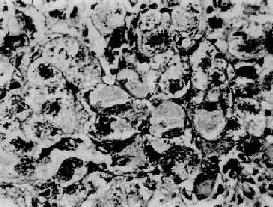
图10-38 毛玻璃样肝细胞
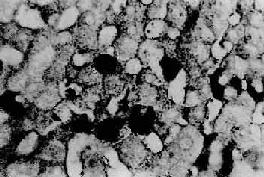
图10-39 肝细胞内的乙型肝炎表面抗原
免疫酶标法(双PAP)染色显示肝细胞浆内的HBsAg
3.重量病毒性肝炎本型病情严重。根据起病急缓及病变程度,可分为急性重型和亚急性重型二种。
(1)急性重型肝炎:少见。起病急,病变发展迅猛、剧烈,病死率高。临床上又称为暴发型、电击型或恶性型肝炎。
本型病变可见肝细胞坏死严重而广泛。肝索解离,肝细胞溶解,出现弥漫性的大片坏死。坏死多自小叶中央开始,向四周扩延,仅小叶周边部残留少数变性的肝细胞。肝窦明显扩张充血并出血,Kupffer细胞增生肥大,并吞噬细胞碎屑及色素。小叶内及汇管区有淋巴细胞和巨噬细胞为主的炎性细胞浸润(图10-40)。残留的肝细胞再生现象不明显。肉眼观,肝体积显著缩小,尤以左叶为甚,重量减至600~800g,质地柔软,表面被膜皱缩(图10-41)。切面呈黄色或红褐色,有的区域呈红黄相间的斑纹状,故又称急性黄色肝萎缩或急性红色肝萎缩。

图10-40 急性重型肝炎
肝细胞大片坏死消失,小叶中心部最重,周边部残存的肝细胞变性。坏死区有炎性细胞浸润

图10-41 急性重型肝炎
肝体积显著缩小,尤以左叶为著,表面被膜皱缩
临床病理联系及结局:由于大量肝细胞的迅速溶解坏死,可导致:①胆红质大量入血而引起黄疸(肝细胞性黄疸);②凝血因子合成障碍导致出血倾向;③肝功能衰竭,对各种代谢产物的解毒功能发生障碍。此外,由于胆红素代谢障碍及血循环障碍等,还可导致肾功能衰竭(肝肾综合征hepatorenal syndrome)。急性重型肝炎的死因主要为肝功能衰竭(肝昏迷),其次为消化道大出血或急性肾功能衰竭等。弥散性血管内凝血(DIC)也较常见,是引起严重出血、致死的另一个因素。
本型肝炎如能渡过急性期,部分病例可发展为亚急性型。
(2)亚急性重型肝炎:多数是由急性重型肝炎迁延而来或一开始病变就比较缓和呈亚急性经过。少数病例可能由普通型肝炎恶化而来。本型病程可达一至数月。
病变特点为,既有大片的肝细胞坏死,又有肝细胞结节状再生。由于坏死区网状纤维支架塌陷和胶原纤维化,致使再生的肝细胞失去原有的依托呈不规则的结节状,失去原有小叶的结构和功能。小叶内外有明显的炎性细胞浸润。小叶周边部小胆管增生并可有胆汁淤积形成胆栓。肉眼观,肝不同程度缩小,被膜皱隔,呈黄绿色(亚急性黄色肝萎缩)。病程长者可形成大小不等的结节,质地略硬。切面黄绿色(胆汁淤积),交错可见坏死区及小岛屿状再生结节。
此型肝炎如及时治疗有停止进展和治愈的可能。病程迁延较长(如1年)者,则逐渐过渡为坏死后性肝硬变。病情进展者可发生肝功能不全。
【发病机制】
病毒性肝炎的发病是病毒与机体之间相互作用的结果。病毒的量、毒力和侵入途径与发病有一定关系。小量病毒往往只引起隐性感染,而大量病毒则可导致严重的病变。肝炎的病变程度和类型与机体的免疫状态也有密切关系。现仅就研究较多的乙型肝炎的肝损伤及其不同类型的发病机制简述如下。
(1)肝细胞损伤的机制:乙型肝炎病毒侵入机体后进入肝细胞内复制繁殖,然后以“发芽”形式从肝细胞释出入血。在肝细胞表面则留下病毒抗原成分,此时并不引起明显的肝细胞损伤。病毒入血后,刺激机体免疫系统产生细胞免疫和体液免疫。前者通过杀伤性T细胞(K细胞)及NK细胞等的直接作用和抗体依赖性的细胞毒作用,后者通过产生各种特异性抗体,均能对血中病毒进行反应加以杀灭。但同时也能对受病毒感染的肝细胞(膜上含病毒抗原成分)进行攻击,使肝细胞受到破坏发生坏死。一般认为,T细胞介导的细胞免疫反应是病毒感染后引起肝细胞损伤的主要因素。近年研究表明,在细胞免疫反应中,靶细胞抗原不一定只是HBsAg,还可有肝细胞膜脂蛋白及HBsAg。
(2)乙型肝炎的发病机制:应用上述肝细胞免疫损伤机制可以解释乙型肝炎出现的不同类型:①T细胞功能正常,感染病毒量多,毒力强时受感染及免疫损伤的肝细胞多而重,表现为急性重型肝炎;②T细胞功能正常,病毒量较少,毒力较弱则发生急性普通型肝炎;③T细胞功能正常,病毒量甚少,毒力很弱则表现为轻型或亚临床型肝炎;④T细胞功能不足,免疫反应仅能清除部分病毒和损伤部分受感染的肝细胞,未清除的病毒可继续繁殖并感染,反复发生部分肝细胞损伤,结果表现为慢性肝炎;⑤机体免疫功能缺陷,T细胞呈免疫耐受状态,此时病毒与宿主共生。病毒在肝细胞内持续复制,感染的肝细胞也不受免疫损伤,此时则表现为无症状的病毒携带者。
四、酒精性肝病
酒精性肝病(alcoholic liver disease)为慢性酒精中毒的主要表现之一。长期大量酗酒者据统计有10%~20%发生此类损伤。严重时出现临床表现,如呕吐、呕血或黑便,其中部分可发生黄疸、肝功能衰竭。
【病变】
慢性酒精中毒主要引起肝的3种损伤,即脂肪肝、酒精性肝炎和酒精性肝硬变。三者可单独出现,也可同时并存或先后移行。一般认为脂肪肝在先,或经过酒精性肝炎再演变为肝硬变,或直接演变为肝硬变。
1.脂肪肝酒精中毒最常见的肝病变是脂肪变性。肝细胞含有相当大的脂滴,可将胞核推挤到细胞一侧,肝细胞肿大变圆。小叶中央区受累明显,有时同时伴有各种程度的肝细胞水样变性。单纯的脂肪肝常无症状。
2.酒精性肝炎(alcoholic hepatitis)在有临床肝症状表现的病例,常出现3种病变:肝细胞脂肪变性,酒精透明小体(alcoholic hyalin,AH 图10-42)形成和灶状肝细胞坏死伴中性粒细胞浸润。
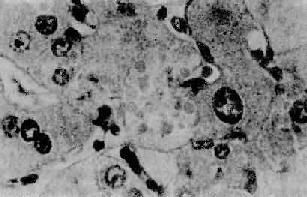
图10-42 酒精性肝炎
图中央区肝细胞浆内见成群的酒精透明小体
3.酒精性肝硬变(alcoholic cirrhosis) 一般认为此种肝硬变是由脂肪肝和酒精性肝炎进展而来。一般的脂肪肝,如继续酗酒则多发展为酒精性炎,再演变为肝硬变。酒精性肝炎时肝细胞发生坏死,最终引起纤维化。相邻肝小叶的纤维化条索相互连接,导致肝小叶的正常结构被分割破坏,发展成假小叶及肝细胞结节状再生,形成酒精性肝硬变。
【发病机制】
肝是酒精代谢、降解的主要场所。酒精对肝有直接损伤作用。其机制如下:
1.NADH对NAD比值增高的效应进入肝内的酒精,在乙醇脱氢酶和微粒体乙醇氧化酶系的作用下转变为乙醛,再转变为乙酸。后一反应使辅酶Ⅰ(NAD)转变为还原型辅酶Ⅰ(NADH),因而NADH/NAD值增高。①NADH增多有抑制线粒体三羧酸循环的作用,从而使肝细胞对脂肪酸的氧化能力降低,可引起脂肪在肝内堆积而发生脂肪肝。②NADH增多可使细胞代谢中的乳酸增多,后者对肝内脂肪变及胶原形成等有促进作用。③NADH增多,使线粒体增加以NADH的再氧化,致耗氧过多。肝细胞因缺氧而易于发生坏死和纤维化。
2.乙醛的毒性作用在酒精代谢过程中产生的乙醛具有强烈的生化反应和毒性,可影响肝细胞膜的性状及抑制肝细胞合成的蛋白的分泌排出。
3.营养缺乏作用嗜酒者常有的营养不足,尤其是蛋白质缺乏,使肝内氨基酸及酶类减少,可以促进酒精的毒性作用。
五、肝硬变
肝硬变(liver cirrhosis)是一种常见的慢性肝病,可由多种原因引起。肝细胞弥漫性变性坏死,继而出现纤维组织增生和肝细胞结节状再生,这三种改变反复交错进行,结果肝小叶结构和血液循环途径逐渐被改建,使肝变形、变硬而形成肝硬变。本病早期可无明显症状,后期则出现一系列不同程度的门静脉高压和肝功能障碍。
肝硬变按病因分类为:病毒肝炎性、酒精性、胆汁性、隐源性肝硬变。按形态分类为:小结节型、大结节型、大小结节混合型及不全分隔型肝硬变(为肝内小叶结构尚未完全改建的早期硬变)。我国常用的分类是结合病因及病变的综合分类,分为:门脉性、坏死后性、胆汁性、淤血性、寄生虫性和色素性肝硬变等。以上除坏死后性相当于大结节及大小结节混合型外,其余均相当于小结节型。其中门脉性肝硬变最常见,其次为坏死后性肝硬变。其它类型较少。
(一)门脉性肝硬变
门脉性肝硬变(portal cirrhosis),旧称雷奈克(Laennec)肝硬变,相当于小结节型肝硬变。为各型肝硬变中最常见者。本病在欧美因长期酗酒者引起多见(酒精性肝硬变),在我国及日本,病毒性肝炎则可能是其主要原因(肝炎后肝硬变)。
【病因和发病机制】
1.病毒性肝炎慢性病毒性肝炎,尤以乙型慢性活动性肝炎是肝硬变的主要原因,其中大部分发展为门脉性肝硬变。在肝硬变患者肝内常显HBsAg阳性。其阳性率高达76.7%。另外,近年明确的丙型肝炎,大部可转为慢性,由慢性活动性肝炎转为肝硬变者达35%。
2.慢性酒精中毒在欧美国家因长期酗酒引起的肝硬变可占总数的40%~50%。
3.营养缺乏此项因素作为肝硬变的原因尚有争议。动物实验表明,饲喂缺乏胆碱或蛋氨酸食物的动物,可经过脂肪肝发展为肝硬变。
4.毒物中毒某些化学毒物对肝有破坏作用,长期作用可引起肝硬变。临床上偶有因含砷的杀虫剂、辛可芬、四氯化碳、黄磷等慢性中毒引起肝硬变的报告。
上述各种因素首先引起肝细胞脂肪变、坏死及炎症等,以后在坏死区发生胶原纤维增生。后者主要来自增生的纤维母细胞、局部的贮脂细胞及因肝细胞坏死,局部的网状纤维支架塌陷,网状纤维融合形成胶原纤维(无细胞硬化)。初期增生的纤维组织虽形成小的条索但尚未互相连接形成间隔而改建肝小叶结构时,称为肝纤维化。为可复性病变,如果病因消除,纤维化尚可被逐渐吸收。如果继续进展,小叶中央区和汇管区等处的纤维间隔互相连接,终于使肝小叶结构和血液循环被改建而形成肝硬变(图10-43)。
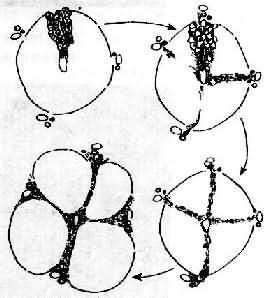
图10-43门脉性肝硬变假小叶形成过程示意图
【病变】
肉眼观,早、中期肝体积正常或略增大,质地正常或稍硬。后期肝体积缩小,重量减轻,由正常的1500g减至1000g以下。肝硬度增加,表面呈颗粒状或小结节状,大小相仿,最大结节直径不超过1.0cm(图10-44)。切面见小结节周围为纤维组织条索包绕。结节呈黄褐色(脂肪变)或黄绿色(淤胆)弥漫分布于全肝。
图10-44 门脉性肝硬变
肝缩小变硬,表面呈弥漫的细颗粒状
镜下,正常肝小叶结构被破坏,由广泛增生的纤维组织将肝小叶分割包绕成大小不等、圆形或椭圆形肝细胞团,即假小叶(图10-45)。假小叶内肝细胞索排列紊乱,小叶中央静脉缺如、偏位或有两个以上,有时还可见被包绕进来的汇管区。再可见再生的肝细胞结节(也可形成假小叶),其特点是肝细胞排列紊乱,胞体较大,核大染色较深,常出现双核肝细胞。增生的纤维组织常压迫、破坏细小胆管,引起小胆管内淤胆。此外,还可见到新生的细小胆管和无管腔的假胆管(已用角蛋白单克隆抗体免疫组化技术证明,假胆管来源于胆管上皮)。
图10-45 门脉性肝硬变
图中央的肝细胞团由增生的结缔组织所包绕,为一个典型的假小叶
由于上述的肝细胞坏死、纤维组织增生和假小叶形成,相应地破坏并改建肝内肝血管系统,导致异常吻合枝的形成和血管网的减少。
【临床病理联系】
1.门脉高压症是由于肝内血管系统在肝硬变时被破坏改建引起:①由于假小叶形成及肝实质纤维化的压迫使小叶下静脉(窦后)、小叶中央静脉及肝静脉窦受压,致门静脉的回流受阻。②肝动脉与门静脉间形成异常吻合支(图10-46),压力高的动脉血流入门静脉,使后者压力增高。
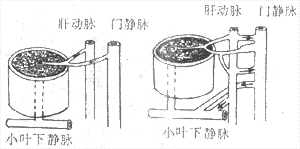
图10-46 肝硬变时肝内血液循环变化示意图
正常时肝内血液循环 肝硬变时肝内血管异常吻合
门脉压升高,胃、肠、脾等器官的静脉血回流受阻。晚期因代偿的调,临床出现:①脾肿大(splenomegaly),重量多在500g以下,少数可达800~1000g。镜下,红髓内有含铁血黄素沉着及纤维组织增生,形成黄褐色的含铁结节。②胃肠淤血,粘膜淤血、水肿,致患者食欲不振,消化不良。③腹水(ascites),在晚期出现,在腹腔内聚积大量淡黄色透明液体(漏出液)。腹水形成原因主要有:由于小叶中央静脉及小叶下静脉受压,肝窦内压上升,液体自窦壁漏出,部分经肝被膜漏入腹腔;肝细胞合成白蛋白功能降低,导致低蛋白血症,使血浆胶体渗透压降低;肝灭能作用降低,血中醛固酮、抗利尿素水平升高,引起水、钠潴留。④侧支循环形成,门静脉压升高使部分门静脉血经门体静脉吻合枝绕过肝直接回心(图10-47)。主要的侧支循环和合并症如:食管下段静脉丛曲张,如破裂可引起大呕血,是肝硬变患者常见的死因之一;直肠静脉丛曲张,破裂常发生便血,长期便血可引起贫血;脐周围静脉网曲张,临床上出现“海蛇头”(caput medusae)现象。
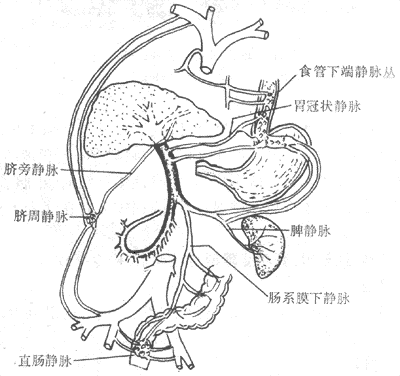
图10-47 肝硬变时侧支循环模式图
2.肝功能不全由于肝实质长期反复受破坏,引起肝功能障碍。主要表现有:①睾丸萎缩,男子乳房发育症(可能因肝对雌激素灭能作用减弱);②蜘蛛状血管痣,为出现于体表的小动脉末梢扩张,也可能与体内雌激素过多有关;③出血倾向,患者有鼻衄、牙龈出血、粘膜、浆膜出血及皮下淤斑等。主要由于肝合成凝血因子及纤维蛋白原减少及脾肿大、功能亢进加强了对血小板的破坏;④肝细胞性黄疸,因肝细胞坏死,胆汁淤积而来,多见于肝硬变晚期;⑤肝性脑病(肝昏迷),是晚期肝功能衰竭引起的一种神经精神综合征,主要由于肠内含氮物质不能在肝内解毒而引起的氨中毒,为肝硬变患者常见的死因之一。
【结局】
肝硬变时肝组织已被增生的纤维组织改建,不易从形态结构上完全恢复正常,但是由于肝有强大代偿能力,只要及时治疗,常使疾病处于相对稳定状态,可维持相当长时期。此时肝细胞的变性、坏死基本消失,纤维母细胞的增生也可停止。但如病变持续进行,发展到晚期,肝功能衰竭,患者可因肝昏迷而死亡。此外,常见的死因还有食管下段静脉丛破裂引起的上消化道大出血,合并肝癌及感染等。
(二)坏死后性肝硬变
坏死后性肝硬变(postnecrotic cirrhosis),相当于大结节型肝硬变和大小结节混合型肝硬变,是在肝实质发生大片坏死的基础上形成的。
【病因】
1.肝炎病毒感染 现知大部为HBV感染,也有HBV与HDV复合感染。VEV的感染多在孕妇。患者多患亚急性重型肝炎,病程迁延数月至一年以上,则逐渐形成坏死后性肝硬变。另外,慢性活动性肝炎反复发作并且坏死严重时,也可引起。
2.药物及化学物质中毒。
【病变】
肉眼观,肝体积缩小,重量减轻,质地变硬。表面有较大且大小不等的结节,最大结节直径可达6cm。由于形成大小不等的结节常使肝变形(图10-48),肝左叶明显萎缩,右叶相对肥大隆起。

图10-48 坏死后性肝硬变
肝体积缩小,表面有大小不等的粗大结节
镜下,肝小叶呈灶状、带状甚至整个小叶坏死,代之以纤维组织增生,形成间隔,将原来的肝小叶分割为大小不等的假小叶。假小叶内肝细胞常有不同程度的变性和胆色素沉着。假小叶间的纤维间隔较宽阔且厚薄不均,其中炎性细胞浸润、小胆管增生均较显著,这些均与门脉型硬变不同。但本型病情进展较慢,病程较久者在病变上亦不易与门脉性硬变鉴别。
较高。
(三)胆汁性肝硬变
胆汁性肝硬变(billiary cirrhosis)是因胆道阻塞淤胆而引起的肝硬变,较少见,可分为继发性与原发性两类。原发性者更为少见。
1.继发性胆汁性肝硬变
病因:常见的原因为胆管系统的阻塞,如胆石、肿瘤(胰头癌、Vater壶腹癌)等对肝外胆道的压迫,引起狭窄及闭锁。在儿童患者多因肝外胆道先天闭锁,其次是总胆管的囊肿、囊性纤维化(cystic fibrosis)等。胆道系统完全闭塞6个月以上即可引起此型肝硬变。
病变:肝体积常增大,表面平滑或呈细颗粒状,硬度中等。肝外观常被胆汁染成深绿或绿褐色。
镜下,肝细胞胞浆内胆色素沉积,肝细胞因而变性坏死。坏死肝细胞肿大,胞浆疏松呈网状、核消失,称为网状或羽毛状坏死。毛细胆管淤胆、胆栓形成。胆汁外溢充满坏死区,形成“胆汁湖”。汇管区胆管扩张及小胆管增生。纤维组织增生使汇管区变宽、伸长,但在较长时期内并不侵入肝小叶内。故小叶的改建远较门脉性及坏死后性肝硬变为轻。伴有胆管感染时则见汇管区及增生的结缔组织内有多量中性粒细胞浸润甚至微脓肿形成。
2.原发性胆汁性肝硬变本病又称慢性非化脓性破坏性胆管炎。很少见,多发生于中年以上妇女,男性患者不超过10%。临床表现为长期梗阻性黄疸、肝大和因胆汁刺激引起的皮肤瘙痒等。但肝内外的大胆管均无明显病变。本病还常伴有高脂血症和皮肤黄色瘤。
病因:不明,一般认为,可能与服用某些药物诱发肝胆管损伤及自向免疫反应有关。
病变:早期汇管区小叶间胆管上皮空泡变性及坏死并有淋巴细胞浸润,其后则有纤维组织的增生及胆小管的破坏、增生并出现淤胆现象。汇管区增生的纤维组织进而侵入肝小叶内,形成间隔,分割小叶最终发展为肝硬变。在汇管区有时可见铜的沉积,系因淤胆造成铜的肠肝循环障碍所致。
(四)其它类型肝硬变
淤血性肝硬变本病见于慢性充血性心力衰竭。长期淤血缺氧,使肝小叶中央区肝细胞陷于萎缩、坏死,最后纤维化。如淤血持续存在,进而形成纤维条索分割肝小叶而形成肝硬变。
色素性肝硬变 多见于血色病(hemochromatosis)患者,由于肝内有过多的含铁血黄素沉着而形成。
寄生虫性肝硬变 主要见于慢性血吸虫病(详见寄生虫病)。
六、药物性肝损伤
进入体内的药物,无论是口服或注射均要经过肝代谢或解毒。某些药物可引起肝损伤。此种损伤系代谢过程中药物或其代谢产物直接对肝的毒性作用所致。损伤程度与药物毒性和剂量有关。药物引起的肝损伤依病变大致可分为3种类型。
1.只引起肝内淤胆、小胆管及毛细胆管胆栓形成并无肝细胞坏死及炎症反应
■[此处缺少一些内容]■
炎时,细菌沿胆道上行蔓延到肝。此种感染较为常见。②血源性感染,主要见于细菌性心内膜炎和脓毒败血症时,此时血中含有的细菌或含菌栓子,经肝动脉入肝引起肝脓肿。③直接感染,如肝开放性创伤或肝周围组织的器官的感染灶直接扩延,以及婴儿的脐带感染等。
感染细菌多数为金黄色葡萄球菌、大肠杆菌、链球菌等。
【病变】
肉眼观,肝体积肿大。初期脓肿较小,直径在1cm左右,球形,多发,散在,黄色。后期互相连通,形成较大的圆形或不规则脓肿(图10-49)。镜下,脓肿腔内为坏死肝组织及脓液。后期可见周围有较多的纤维肉芽组织增生形成脓肿壁。
图10-49 细菌性肝脓肿
肝切面上见多发性脓肿灶,其大小不等,主要位于肝右叶
肝脓肿靠近膈面者,可穿破肝表面而形成膈下脓肿;穿破膈可向上至胸腔形成脓胸及肺脓肿;穿破心包形成心包积脓等。
(二)阿米巴性肝脓肿
阿米巴性肝脓肿并非真性脓肿,而是阿米巴滋养体的溶组织酶引起的肝组织液化性坏死。这是肠阿米巴痢疾最常见的并发症。多发生在肝右叶。早期为小的病灶,以后逐渐发展为单一的大脓肿,内含咖啡色半液体状果酱样液化坏死组织。在脓肿外周部分,未完全坏死的肝实质及间质成分常呈破棉絮状。镜下,在坏死与正常组织交界处,有较多的阿米巴滋养体及少量单核细胞浸润,炎症反应轻微。
八、胆石症
在胆道系统中,胆汁的某些成分(胆色素、胆固醇、粘液物质及钙等)可以在各种因素作用下析出、凝集而形成结石。发生于各级胆管内的结石称胆管结石,发生于胆囊内的结石称胆囊结石,统称胆石症(cholelithiasis)。
【胆石的种类和特点】
按组成成分可将胆石分为色素石、胆固醇石及混合石三种基本类型(图10-50)。
图10-50 各种胆石模式图
左上及右上为混合性结石(表面及剖面)中央为胆色素性结石 左下及右下为胆固醇性结石剖面
1.色素性胆石结石成分以胆红素钙为主,可含少量胆固醇。有泥沙样及砂粒状二种。砂粒状者大小为1~10mm,常为多个。多见于胆管。
2.胆固醇性胆石结石的主要成分为胆固醇。此类结石在我国较欧美为少,其发生率大约不超过胆石症的20%。结石呈圆形或椭圆形。黄色或黄白色,表面光滑或呈细颗粒状,质轻软。剖面呈放射状。多见于胆囊,常为单个,体积较大,直径可达数厘米。
3.混合性胆石由二种以上主要成分构成。以胆红素为主的混合性胆石在我国最多见,约占全部胆石症病例的90%以上。结石多为多面体,少数呈球形,呈多种颜色。外层常很硬,切面成层,或像树干年轮或呈放射状。多发生于胆囊或较大胆管内,大小、数目不等,常为多个,一般约20~30个。
我国胆石症的特点:①胆石类型。混合性色素泥沙样结石远多于胆固醇性结石。②发病部位。胆管多于胆囊,肝内胆管结石发生率也较高。③病因。既往统计胆道蛔虫症在胆石形成上起重要作用。近年由于饮食卫生及营养水平的不断提高已有所改变。
【原因及机制】
胆石形成的基本因素有:胆汁理化状态的改变;胆汁淤滞;感染等三种。常为二种以上因素联合致病。
1.胆汁理化性状的改变由于胆汁理化性状的变化,使其中胆色素或胆固醇析出,形成结石。
2.胆汁淤滞使胆汁中水分被过多吸收,胆汁过度浓缩,使胆色素浓度增高,胆固醇过饱和等,都可促进胆石形成。
3.细菌感染胆道感染时的炎性水肿、细胞浸润和慢性期的纤维增生可使胆道壁增厚,胆道狭窄乃至闭塞,从而引起胆汁淤滞。炎症时渗出的细胞或脱落上皮、细菌群、蛔虫残体及虫卵等也可作为结石的核心,促进胆石形成。
胆固醇性结石的形成机制胆汁中如胆固醇含量过多呈过饱和状态,则易析出形成胆固醇结石(如长期进食高脂肪饮食)。正常时胆汁中一定浓度的胆盐和卵磷脂可以和胆固醇、蛋白质组成混合体胶粒,混悬于胆汁中而不析出。在某些肠疾病时由于丧失了胆盐则促进胆固醇的析出,形成结石。
胆红素性结石形成机制胆汁中的游离胆红素浓度增高可与胆汁中的钙结合,形成不溶性的胆红素钙而析出。正常胆汁中的胆红素多葡萄糖醛酸结合成酯类而不游离。大肠杆菌等肠道细菌中的葡萄糖醛酸酶则有分解上述酯类使胆红素游离出来的作用。所以有肠道菌感染的肠道蛔虫症及胆道炎时易形成胆红素结石。此外,胆色素含量增加(如红细胞破坏过多),胆汁内钙量增加以及胆汁的酸度增加等都可促进此类结石的形成。
九、胆管炎和胆囊炎
胆道炎症主要累及胆管者称胆管炎(cholangitis);主要累及胆囊者称胆囊炎(cholecystitis)。两者又各分为急性和慢性两种。
【病因】
本病多由细菌引起,且多有胆汁淤滞作为发病的基础。淤胆时,胆汁理化状态发生变化可刺激胆道粘膜使其抵抗力降低。主要感染的细菌为大肠杆菌、副大肠杆菌、葡萄球菌等。入侵和细菌可经淋巴道或血道到达胆道,也可以由肠腔经十二指肠乳头逆行进入胆道,后者在我国更为多见。
【病变】
1.急性胆管炎和胆囊炎两者病变相同。粘膜充血水肿,上皮细胞变性、坏死脱落,管壁内不同程度的中性粒细胞浸润。在胆囊者常伴有粘膜腺分泌亢进(卡他性胆囊炎)。如机体抵抗力强或及时治疗,炎症可吸收消退。如病变继续发展,胆囊壁各层均为白细胞弥漫浸润(蜂窝织炎性胆囊炎),浆膜面常有纤维素脓性渗出物覆盖。如胆囊管阻塞,可引起胆囊积脓。如因痉挛、水肿、梗阻及淤胆等导致胆管或胆囊壁的血液循环障碍时,该处可发生出血坏死(坏疽性胆囊炎),甚至发生穿孔,引起胆汁性腹膜炎。
2.慢性胆管炎和胆囊炎多由急性者反复发作迁延而来。此时胆管及胆囊粘膜多发生萎缩,各层组织中均有淋巴细胞、单核细胞浸润和明显纤维化。有时管壁因水肿、纤维增生性肥厚而致管道狭窄。慢性胆囊炎时因囊壁受反复炎性损害,在修复过程中粘膜上皮向囊壁内凹陷生长,有时深达肌层,形成Rokitansky-Aschoff窦。此种现象可见于约90%的慢性胆囊炎病例。在此基础上腺上皮有时可发生癌变(胆囊癌)。
十、原发性肝癌
原发性肝癌(primary carcinoma of liver)是由肝细胞或肝内胆管上皮细胞发生的恶性肿瘤,简称肝癌。其发生率在各国和地区差异很大,在亚非国家较常见,我国发病率较高,属于常见肿瘤之一。发病年龄多在中年以上,男多于女。近年来,我国对肝癌的防治研究取得了显着的成绩。一些直径在1cm以下的早期肝癌(小肝癌)已被发现并取得满意的疗效。对诊断肝癌有辅助价值的甲胎蛋白(AFP)测定已广泛应用,肝癌时其阳性率占70%以上。
【病变】
肉眼类型 早期肝癌或小肝癌是指瘤体直径在3cm以下,不超过2个瘤结节的原发性肝癌。瘤结节呈球形或分叶状,灰白色质较软,切面无出血坏死,与周围组织界限清楚(图10-51)。晚期肝癌,肝体积明显增大可达2000g以上。癌组织可局限于肝的一叶(多为右叶),也可弥散于全肝并大多合并肝硬变。有时硬变的再生结节与癌结节肉眼不易区别。肉眼可分三型。

图10-51 小肝癌伴肝硬变
图为切除的部分肝组织,右侧见一圆形、直径约3cm的单一癌结节
1.巨块型肿瘤为一实体巨块,有的可达儿头大,圆形,多位于肝右叶内甚至占据整个右叶(图10-52)。瘤块质地较软,中心部常有出血坏死。瘤体周边常有散在的卫星状瘤结节。不合并或合并轻度的肝硬变。
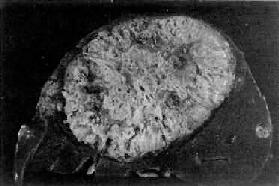
图10-52 原发性肝癌(巨块型)
中央为巨大的灰白色癌块,外围可见散在的小型癌结节
2.多结节型最多见,常发生于较重肝硬变的肝内。瘤结节多个散在,圆形或椭圆形,大小不等,直径由数毫米至数厘米,有的相互融合形成较大的结节(图10-53)。被膜下的瘤结节向表面隆起致肝表面凹凸不平。
图10-53 原发性肝癌(结节型)
肝切面见多数圆形、椭圆形、大小不等的癌结节
3.弥漫型癌组织在肝内弥漫分布,无明显的结节或形成极小结节。常发生在肝硬变基础上,少见。
组织学类型:过去认为组织学类型对预后评估意义不大,近年研究表明,有些特殊类型,如透明细胞型、层状纤维化型(fibrolamellar type)及微小或包裹型(minute or encapsulated type)肝癌预后均较好。按组织发生可将肝癌分为三大类。
1.肝细胞癌最多见,是由肝细胞发生的肝癌。其分化较好者癌细胞类似肝细胞。分化差者癌细胞异型性明显,常有巨核及多核瘤细胞。有的癌细胞排列成条索状(索状型)(图10-54);亦可呈腺管样(假腺管型)或实体团块状(实体型)。有时癌细胞为深染的小细胞(小细胞型)。有时癌组织中有大量纤维组织分割(硬化型),分割的纤维组织多且呈分层状(层状纤维化型)。
图10-54 原发性肝细胞癌
左下:肝细胞区 右上:坏死崩解的癌细胞 中间区:索状型肝细胞癌,癌细胞有明显的异型性
2.胆管上皮癌较为少见,是由肝内胆管上皮发生的癌。其组织结构多为腺癌或单纯癌。较少合并肝硬变。有时继发于华支睾吸虫病。
3.混合性肝癌具有肝细胞癌及胆管上皮癌两种结构,最少见。
肝母细胞瘤(hepatoblastoma) 是婴、幼儿原发的肝恶性肿瘤,患者常合并大肠息肉病。AFP反应常呈阳性。肉眼观为界限清楚的孤立性肿块,多位于肝右叶。大小5~25cm不等。瘤组织常有囊性变、坏死及出血。镜下,瘤组织包含有分化程度不同的上皮及间叶成分。上皮性瘤细胞有小梭形、深染少胞浆的胚胎型细胞及较大有胞浆的胎儿型细胞。此细胞有时胞浆含糖原呈透明或颗粒状。间叶成分中可见有骨样、软骨样组织及分化为肌组织。
【蔓延和转移】
肝癌首先在肝内蔓延和转移。癌细胞常沿门静脉播散,在肝内形成转移癌结节,还可逆行蔓延至肝外门静脉主干,形成较大的癌栓,有时可阻塞管腔引起门静脉高压。肝外转移常通过淋巴道转移至肝门淋巴结、上腹部淋巴结和腹膜后淋巴结。晚期可通过肝静脉转移到肺、肾上腺、脑及骨等处。有时肝癌细胞可直接种植到腹膜和卵巢表面,形成种植性转移。
【临床病理联系】
临床上多有肝硬变病史,进行性消瘦,肝区疼痛、肝迅速增大,黄疸及腹水等表现。有时由于肝表面癌结节自发性破裂或侵破大血管而引起腹腔内大出血。由于肿瘤压迫肝内外胆管及肝组织广泛破坏而出现黄疸。
【病因】
以下因素与肝癌发生有关。
1.病毒性肝炎现知乙型肝炎与肝癌有密切关系,其次为近年确定的丙型肝炎。肝癌病例HBsAg阳性率可高达81.82%。国外报告,在肝癌高发地区有60%~90%的肝癌来自HBV。近年报道,在HBV阳性的肝癌患者可见HBV基因整合到肝癌细胞DNA中。因之认为HBV是肝癌发生的重要因素。最近,HCV的感染也被认为可能是肝癌发生的病原因素之一。据报道,在日本有70%,在西欧有65%~75%的肝癌患者发现HCV抗体阳性。
2.肝硬变 肝硬变与肝癌之间有密切关系。据统计,一般需经7年左右肝硬变可发展为肝癌。其中以坏死后性肝硬变为最多,肝炎后肝硬变次之,门脉性肝硬变最少。
3.真菌及其毒素黄曲霉菌、青霉菌、杂色曲霉菌等都可引起实验性肝癌。其中以黄曲霉菌(aspergillus flavus)最为重要。用该菌或其毒素(aflatoxin),或被其污染的食物均可诱发动物肝癌。在肝癌高发区,食物被黄曲霉菌污染的情况往往也较严重。
4.亚硝胺类化合物从肝癌高发区南非居民的食物中已分离出二甲基亚硝胺。此类化合物也可引起其它处肿瘤如食管癌。
十一、胆道癌
(一)肝外胆管癌
肝外胆管癌通常为腺癌,少数为鳞化腺癌及鳞癌等。长期慢性炎症、结石及囊性扩张等与癌发生可能有关。临床常表现为右上腹疼痛、包块、黄疸等。
胆管癌可发生于胆管的任何部位,在左右肝管分叉部最多见。肉眼观可分为绒毛状、结节性及弥漫型。镜下绝大多数为腺癌,包括乳头状腺癌、粘液性腺癌等;少数为腺鳞癌或鳞癌。也见有硬癌。
(二)胆囊癌
胆囊癌少见,但预后较差。女性较多见。多发生于长期患胆囊良性病变者,尤其是胆石症、慢性胆囊炎等。
胆囊癌多发生于胆囊颈部。组织学类型,90%为分化较高的腺癌,少数为腺鳞癌或鳞癌。在腺癌中约半数以上为硬癌,其次为乳头状腺癌,粘液癌及未分化癌少见,类癌罕见。肉眼观,胆囊癌多呈弥漫浸润性生长,使囊壁增厚,变硬,灰白色,砂粒样。粘膜无明显肿块,与慢性炎症或瘢痕不易区别。有时呈息肉状生长,基底部较宽。胆囊床及邻近肝组织内常有转移灶形成。邻近器官如十二指肠、结肠和胃等亦可发生转移。此外,还可发生胆囊管、局部淋巴结及小网膜淋巴结的转移。血行转移少见。
第四节 胰腺疾病
一、胰腺炎
胰腺炎(pancreatitis)是胰腺因胰蛋白酶的自身消化作用而引起的疾病。可分为急性及慢性二种。
【病因和发病机制】
本病主要由胰腺组织受胰蛋白酶的自身消化作用。在正常情况下,胰液内的胰蛋白酶原无活性,待其流入十二指肠,受到胆汁和肠液中的肠激酶(enterodinase)的激活作用后乃变为有活性的胰蛋白酶,方具有消化蛋白质的作用。胰腺炎时因某些因素(下述)激活了胰蛋白酶,后者又激活了其它酶反应,如弹性硬蛋白酶(elastase)及磷脂酶A(phospholipase A),对胰腺发生自身消化作用,促进了其坏死溶解。已查出在胰腺腺泡的酶原颗粒中含有高浓度的弹性硬蛋白酶,在胰腺分泌液中含有无活性的该酶前体,后者可被胰蛋白酶激活而能溶解弹性组织,从而破坏血管壁及胰腺导管。另外,胰蛋白酶对由脂蛋白构成的细胞膜及线粒体膜并无作用,而胰液中的磷脂酶A被脱氧胆酸激活后,作用于细胞膜和线粒体膜的甘油磷脂,使之分解变为脱脂酸卵磷脂,亦称溶血卵磷脂(lysolecithin),后者对细胞膜有强烈的溶解作用,可溶解、破坏胰腺细胞膜和线粒体膜的脂蛋白结构,致细胞坏死。脂肪坏死也同样先由胰液中的脱脂酸卵磷脂溶解、破坏了脂肪细胞膜后,胰脂酶才能发挥作用。
急性胰腺炎时胰酶被激活的原因概括如下。
1.十二指肠壶腹部的阻塞引起胆汁返流(biliary reflux)总胆管和胰管共同开口于十二指肠壶腹部,返流的胆汁可进入胰管(共道说),将无活性的胰蛋白酶原激活成胰蛋白酶,再诱发前述一系列酶反应引起胰腺的出血、坏死。引起十二指肠壶腹部阻塞的原因有胆石、蛔虫、暴饮暴食引起的壶腹括约肌痉挛及十二指肠乳头水肿等。后二种原因也可使十二指肠液进入胰内。
2.胰液分泌亢进使胰管内压升高暴饮暴食,酒精的刺激使胃酸及十二指肠促胰液素secretin分泌增多,进而促进胰液分泌增多,造成胰管内压增高。重者可导致胰腺小导管及腺泡破裂,放出内生性活素,激活胰蛋白酶原等,从而引起胰腺组织的出血坏死。
在急性胰腺炎的实际发病上很可能是上述两种因素的综合作用,即胰液分泌亢进和不全阻塞并存。近年又注意到受细菌感染的胆汁可破坏胰管表面被覆的粘液屏障,强调了胆道感染在本病发生上的重要性。
(一)急性胰腺炎
急性胰腺炎是胰酶消化胰腺及其周围组织所引起的急性炎症,主要表现为胰腺呈炎性水肿、出血及坏死,故又称急性出血性胰腺坏死(acute hemorrhagic necrosis of pancreas),好发于中年男性,发作前多有暴饮暴食或胆道疾病史。临床表现为突然发作的上腹部剧烈疼痛并可出现休克。
【病变】
按病变表现不同,可将本病分为急性水肿性(或间质性)胰腺炎及急性出血性胰腺炎二型。
1.急性水肿性(间质性)胰腺炎较多见,约占急性胰腺炎全部病例的3/4或更多。病变多局限在胰尾。病变的胰腺肿大变硬,间质充血水肿并有中性粒及单核细胞浸润。有时可发生局限性脂肪坏死,但无出血。本型预后较好,经治疗后病变常于短期内消退而痊愈。少数病例可转变为急性出血性胰腺炎。
2.急性出血性胰腺炎较少见。本型发病急剧,病情及预后均较水肿型严重。病变以广泛的胰腺坏死、出血为特征,伴有轻微炎症反应。
肉眼观,胰腺肿大,质软,出血,呈暗红色,分叶结构模糊。胰腺、大网膜及肠系膜等处散在混浊的黄白色斑点状或小块状的脂肪坏死灶。坏死灶是由于胰液溢出后,其中的脂酶将中性脂肪分解成甘油及脂肪酸,后者又与组织液中的钙离子结合成不溶性的钙皂而形成。
镜下,胰腺组织呈大片凝固性坏死,细胞结构模糊不清,间质小血管壁也有坏死,这是造成胰腺出血的原因。在坏死的胰腺组织周围可见中性及单核细胞浸润。患者如渡过急性期,则炎性渗出物和坏死物逐渐被吸收,局部发生纤维化而痊愈或转变为慢性胰腺炎。
【临床病理联系】
1.休克患者常出现休克症状。引起休克的原因可有多种,如由于胰液外溢,刺激腹膜引起剧烈疼痛;胰腺组织及腹腔内出血;组织坏死,蛋白质分解引起的机体中毒等。休克严重者抢救不及时可以致死。
2.腹膜炎由于急性胰腺坏死及胰液外溢,常引起急性腹膜炎。
3.酶的改变胰腺坏死时,由于胰液外溢,其中所含的大量淀粉酶及脂酶可被吸收入血并从尿中排出。临床检查常见患者血清及尿中淀粉酶及脂酶含量升高,可助诊断。
4.血清离子改变患者血中的钙、钾、钠离子水平下降。血钙下降的原因,近年研究认为急性胰腺炎时胰腺α细胞受刺激,分泌胰高血糖素(glucagon),后者能使甲状腺分泌降钙素,抑制钙自骨质内游离,致使胰腺炎时因脂肪坏死而消耗的钙得不到补充而发生血钙降低。血钾、钠的下降可能因持续性呕吐造成。
(二)慢性胰腺炎
慢性胰腺炎是由于急性胰腺炎反复发作造成的一种胰腺慢性进行性破坏的疾病。有的病例急性期不明显,症状隐匿,发现时即属慢性。临床上常伴有胆道系统疾患,患者有上腹痛、脂性泻,有时并发糖尿病。慢性酒精中毒时也常引起本病。
【病变】
肉眼观,胰腺呈结节状,质较硬。切面可见胰腺间质纤维组织增生,胰管扩张,管内偶见有结石形成。有时可见胰腺实质坏死,坏死组织液化后,被纤维组织包围形成假囊肿。镜下,可见胰腺小叶周围和腺泡间纤维组织增生或广泛纤维化,腺泡和胰腺组织萎缩、消失,间质有淋巴细胞、浆细胞浸润。
二、胰腺癌
胰腺癌(carcinoma of pancreas)在消化系统癌中较为少见,据不完全统计,在我国占人体癌的1%。日本统计,在70年代以后有明显上升趋势,这可能与饮食中蛋白质、脂肪成分逐年增高有关。患者年龄多在40~70岁之间,男多于女。
【病变】
胰腺癌可发生于胰腺的头、体、尾部或累及整个胰腺,但以胰头部最多,约占胰腺癌的60%~70%。Bell通过609例尸检统计,本癌发生在胰头部者占59%,体部18%,尾部7.5%,弥漫性者15%。胰腺癌的大小和外形不一,呈圆形或卵圆形。边界有的分明,有的弥漫浸润与邻近胰腺组织难以分辨。有的呈凹陷硬块状埋在胰腺之中,活检时需深取组织方能查出。有的甚至在开腹探查时,肉眼上仍很难与慢性胰腺炎时增粗变硬的胰腺相鉴别。
镜下,胰腺癌有以下几种类型
1.腺癌癌细胞来自导管上皮,排列成腺样,有腔,称腺管型腺癌(tubular adenocarcinoma),此型占大部分,约80%,其中有的为乳头状腺癌或乳头状囊腺癌;也有的来自腺泡称腺泡型腺癌(acinar adenocarcinoma)。分化较高的病例可见癌细胞呈楔形或多角形,胞浆含嗜酸性颗粒,形成腺泡或小团块,极似正常胰腺腺泡。有时腺癌细胞产生粘液,胞浆透明。
2.单纯癌癌细胞呈圆形或多角形,有时可出现多形性癌细胞或癌巨细胞,无腺体结构。有的癌细胞形成细条索状胞巢,被间质结缔组织分隔,呈硬癌构型。有的癌细胞多,间质少,形成髓样癌,这样的癌组织易发生坏死而形成囊腔。
3.鳞状细胞癌少见,来自胰腺管上皮的鳞状化生。有时可见鳞状细胞癌和产生粘液的腺癌合并发和生(腺鳞癌)。
【扩散转移】
胰头癌早期可直接蔓延到邻近组织如胆管与十二指肠,稍后即转移至胰头旁及总胆管旁淋巴结,肝内也可有少数转移灶。胰体癌及尾部癌的扩散则较为广泛。常直接与腹腔神经丛接触并侵入神经周围淋巴间隙,进一步发生远隔部位的淋巴道或血道转移,常在肺内形成转移癌。
【临床病理联系】
胰头癌早期可有总胆管梗阻现象,临床上出现逐渐加重的黄疸。胰体尾部癌临床上常无黄疸,但常因癌组织侵入门静脉而产生腹水,压迫脾静脉发生脾肿大,侵入腹腔神经丛而发生深部疼痛。有时患者血清中胰蛋白酶、亮氨酸氨基肽酶(leucine aminopeptidase)、淀粉酶等均增高,但由于其结果均不恒定,在临床诊断上不常应用。有报告约80%~90%患者血清CEA检出阳性,但因CEA的特异性不强,亦不能作为诊断本癌的根据。
第十一章 造血系统疾病
造血系统包括造血器官和血液。胚胎时期肝、骨髓、脾、淋巴结等都参与造血过程。出生后主要的造血器官为骨髓。在疾病或骨髓代偿功能不足时,肝、脾、淋巴结可恢复胚胎时期的造血功能称为髓外造血。传统习惯常将造血器官和组织分为髓样组织(myeloid tissue)和淋巴组织。髓样组织包括骨髓和骨髓中所产生的各种细胞,即红细胞、血小板、料细胞和单核细胞。淋巴组织包括胸腺、脾、淋巴结和分散的淋巴组织。实际上这两种组织并不能截然分开,例如成熟的淋巴细胞多不在骨髓内,但淋巴干细胞则由骨髓产生;白细胞的恶性肿瘤——白血病来源于骨髓但常累及淋巴结和脾。造血系统功能重要,淋巴细胞和单核细胞并属于免疫系统,对机体有重要的防御作用。机体内外环境中的刺激因素都能引起这些细胞和组织的反应,产生相应的变化。造血系统的疾病种类繁多,包括造血系统各种成分的量和质的变化。本章重点叙述几种常见的造血系统增生性疾病和肿瘤。
第一节 淋巴结反应性增生
淋巴结是机体重要的免疫器官。各种损伤和刺激常引起淋巴结内的淋巴细胞和组织细胞反应性增生,使淋巴结肿大,称为淋巴结反应性增生。其原因很多,包括细菌、病毒、毒物、代谢的毒性产物、变性的组织成分及异物等,都可成为抗原或致敏原刺激淋巴组织引起反应。淋巴结肿大的程度不等,有时可达10cm。镜下,由于致病原因不同,淋巴结反应性增生的成分和分布情况不同。刺激B细胞的抗原物质主要引起淋巴滤泡增生增大,生发中心扩大增生;刺激T细胞的抗原物质主要引起滤泡旁区淋巴细胞增生。有些抗原物质则主要引起淋巴窦内的组织细胞增生。淋巴结反应性增生为良性病变,但肿大的淋巴结无论肉眼观或镜下都容易与淋巴结的肿瘤混淆,但其治疗和预后差别很大,应注意鉴别。
淋巴结增生的类型较多,现简述如下:
一、非特异性反应性淋巴滤泡增生
非特异性反应性淋巴滤泡增生(nonspecific reactive follicular hyperplasia)的主要特点为淋巴结肿大,淋巴滤泡增生,生发中心明显扩大。淋巴滤泡数量增多,不仅分布于淋巴结皮质,并可散在于皮髓质交界处和髓质内。滤泡大小形状不一,界限明显。生发中心明显扩大、增生,内有多数各种转化的淋巴细胞,核较大,有裂或无裂,核分裂像多见,并有多数吞噬细胞,胞浆内含有吞噬的细胞碎屑。生发中心周围有小淋巴细胞环绕。在滤泡之间的淋巴组织内可见浆细胞、组织细胞及少数中性粒细胞和嗜酸性粒细胞浸润。淋巴窦内的网状细胞和内皮细胞增生。
反应性淋巴滤泡增生易与滤泡性淋巴瘤混淆,后者的淋巴结结构破坏,滤泡大小形状相似,界限不明显。滤泡内增生的细胞呈异型性,但类型比较一致,核分裂像较少,一般不见吞噬异物的巨噬细胞,增生的淋巴细胞为单克隆性;而反应性淋巴滤泡增生时增生的淋巴细胞为多克隆性。
二、巨大淋巴结增生
巨大淋巴结增生(giant lymph node hyperplasia)又称血管滤泡性淋巴结增生(angiofollicular lymph node hyperplasia)或Castleman淋巴结增生症(lymph node hyperplasia of Castleman;Castleman disease)。这是一种特殊类型的淋巴结增生,不是肿瘤也不是错构瘤。可发生于任何年龄。
巨大淋巴结增生最常发生于纵隔淋巴结,也可见于肺门淋巴结及颈部、腋窝、肠系膜、阔韧带和腹膜后淋巴结。淋巴结明显肿大,大的直径3~7cm,可达16cm,常呈圆形,包膜完整,界限清楚,切面呈灰白色。
【镜下可分为两种亚型】
玻璃样-血管型(hyaline-vascular type):最多见。巨大淋巴结增生中约90%以上属于这种类型。患者多无症状。淋巴结内淋巴滤泡增生,散在于淋巴结皮质和髓质内。一般淋巴滤泡和生发中心不大。淋巴结内毛细血管增生伸入淋巴滤泡。这些毛细血管内皮细胞肿胀,血管周围常有胶原纤维或玻璃样物质环绕,位于淋巴滤泡中央很象胸腺的Hassall小体。多数成熟的小淋巴细胞在生发中心周围呈向心性排列成葱皮样层状。滤泡之间的淋巴组织中也有多数血管,血管周围有纤维组织或胶原纤维环绕,并常伴有浆细胞、免疫母细胞、嗜酸性粒细胞和组织细胞浸润。有些病例增生的淋巴滤泡主要由小淋巴细胞组成,只有少数滤泡内有小生发中心,称为淋巴细胞型。这种类型最容易与滤泡性淋巴瘤混淆。
浆细胞型(plasma cell type):较少,约占10%。患者常伴有全身症状,如发热、乏力、体重减轻、贫血、红细胞沉降率升高、血液丙种球蛋白增高和低白蛋白血症。淋巴结切除后症状可消失。
淋巴结内淋巴滤泡增生,生发中心明显扩大,周围的淋巴细胞较少。生发中心内各种细胞增生,核分裂像多见,并有许多吞噬了细胞碎屑的巨噬细胞。但中央没有血管,也没有玻璃样变物质。淋巴滤泡之间有大量浆细胞,其间也可有较少数淋巴细胞、免疫细胞和组织细胞浸润。
有些病人在同一淋巴结内二种亚型的变化可同时存在。因此有些作者认为这二种亚型可能为一个过程的不同阶段。浆细胞型可能是早期病变,以后发展为玻璃样-血管型。
三、血管免疫母细胞性淋巴结病
血管免疫母细胞性淋巴结病(angioimmunoblastic lymphadenopathy)又称免疫母细胞性淋巴结病(immunoblastic lymphadenopathy),多发生于中、老年人。主要表现为发热,体重减轻,全身淋巴结肿大,肝、脾肿大,皮肤斑丘疹,瘙痒,并常有多克隆性高丙种球蛋白血症和溶血性贫血。
免疫母细胞性淋巴结病的主要病变为全身淋巴结肿大,淋巴结直径一般约为2~3cm,灰白色,质软,活动,有时有压痛。镜下可见淋巴结的结构消失,淋巴滤泡和淋巴窦不明显。淋巴结内有大量免疫母细胞和转化的淋巴细胞浸润。有时还有多数浆细胞、嗜酸性粒细胞、巨噬细胞和上皮样细胞。毛细血管后小静脉明显增生呈分支状。血管内皮细胞肿胀增生。淋巴结间质内可见无定形伊红色蛋白样物质沉积。除淋巴结外,肝、脾、骨髓和肺内也可见类似病变。
这种疾病的原因和性质尚不清楚。有些病人在上呼吸道病毒感染后发病,有些病人发病前有用某些药物如抗菌素的病史。过去曾认为本病是T细胞调节障碍引起的B细胞过度增生。近年来的研究发现,在很多病人是T细胞淋巴瘤。基因重组研究证实为单克隆性T细胞增生。目前认为本病可能是免疫系统功能障碍引起的克隆性淋巴细胞增生,在此基础上出现恶性细胞株大量增生,发展成为恶性淋巴瘤。
本病预后差别很大。约半数病人不经任何治疗可生存2~4年,约25%用激素治疗或并用其他化疗药物可缓解。有些病变为进行性,可发展为恶性淋巴瘤,预后不佳。到晚期由于免疫功能低下,多数病人死于继发感染。
第二节 组织细胞增生症X
组织细胞增生症 X(histiocytosis X)包括Letterer-Siwe病,Hand-Schüller-Christian病和骨的嗜酸性肉芽肿(eosinophilic granuloma)3种疾病。这三种疾病的临床表现和病理变化不同。Letterer-Siwe病为急性弥漫性,发展快,常表现为恶性过程;Hand-Schüller-Christian病为慢性进行性疾病;嗜酸性肉芽肿为良性局限性组织细胞增生。这三种疾病的病变范围和预后不同。它们的共同特点是都有一种特殊的组织细胞即Langerhans细胞增生。
Langerhans细胞属于单核吞噬细胞系统,来源于骨髓,由单核细胞转化而来。Langerhans细胞正常时分布于皮肤、口腔、食管和阴道粘膜,是一种树突状细胞,散在于上皮细胞之间,也存在于淋巴结、胸腺和脾等处。其直径约12μm,胞浆丰富,染伊红色,核形状不规则,有切迹或分叶状。Langerhans细胞具有CD1,HLA-DR和S-100蛋白等免疫标记。电镜下可见胞浆内有一特殊的细胞器称为Langerhans小体或Birbeck颗粒。这是一种呈杆状的管状结构,中央有一纵形条纹和横行平行排列的周期性条纹,形似一条小拉链。有时一端有泡状膨大似网球拍状。Birbeck颗粒是Langerhans细胞所特有的超微结构特征。
组织细胞增生症X时,增生组织细胞的表型、细胞化学和功能等都与Langerhans细胞非常相似,因此目前认为组织细胞增生症X是Langerhans细胞或其前身的增生性疾病,故称之为Langerhans细胞性组织细胞增生症(Langerhans cell histiocytosis)。
一、Letterer-Siwe病
Letterer-Siwe病又称急性弥散性组织细胞增生症(acute disseminated histiocytosis)是一种急性进行性全身性疾病,主要引起皮肤、内脏器官和广泛的骨组织损害,多见于3岁以下的婴幼儿,3岁以上的儿童中很少见,很少发生于成人。发病急,进展快,病程短。主要症状为发热、皮疹及肝、脾和全身淋巴结肿大。常伴有进行性贫血、粒细胞和血小板减少。
【病理变化】
主要病变为全身性Langerhans细胞增生。增生的Langerhans细胞多数分化较好,有些分化不成熟,体积大,大小、形状不等。胞浆丰富呈伊红色,核椭圆或肾形,常有纵行沟纹呈分叶状,核分裂像很少。有些增生的细胞可吞噬含铁血黄素,有少数吞噬红细胞,后期可吞噬脂类,故胞浆呈空泡状。同时可有少量嗜酸性粒细胞、巨噬细胞、浆细胞和淋巴细胞浸润,有时可见多核巨细胞。
病变范围很广,可累及全身各器官和组织,但以皮肤、脾、淋巴结和骨组织最为常见。皮肤病变多发生于胸背部,四肢较少。早期皮肤上出现少数红色或棕色小丘疹,以后可发展为广泛的斑丘疹或多数小结节,表面呈鳞片状,常溃破、出血。镜下可见皮肤表皮萎缩,过度角化,真皮表层有大量Langerhans细胞浸润。
淋巴结、肝和脾均明显肿大,切面可有出血和坏死区。淋巴窦、肝窦和脾红髓内Langerhans细胞大量增生呈弥漫性浸润,并可逐渐浸润邻近组织。
骨组织损害多发生于颅骨、盆骨和长骨。骨组织中有局限性的暗红色或灰红色肿块,骨组织破坏,再生不明显。X线检查见骨质缺损。骨髓腔内可有大量成片的Langerhans细胞增生,胞浆内常含有多少不等的脂质。
肺也常被累及,增生的Langerhans细胞在肺组织内浸润形成结节状病灶,类似粟粒性结核,以后可引起肺纤维化和肺气肿。
Letterer-Siwe病预后与发病年龄有关,小于2岁者病变进展很快,如无有效治疗常在数周或数月内由于严重贫血和全血细胞减少,并发感染或出血而死亡。
二、Hand-Schüller-Christian病
Hand-Schüller-Christian病又称慢性进行性组织细胞增生症(chronic progressive histiocytosis)。病程较长。多见于幼儿,一般多在2~6岁发病,也可见于青年人,大多数在30岁以下,年长者较少。病变为多发性,主要累及骨骼。皮肤、肺、肝、脾、淋巴结等也可受累。病人常有发热、皮疹、上呼吸道感染、轻度淋巴结肿大和肝、脾肿大。
【病理变化】
病变分布广泛,以骨组织最为突出,多发生在颅骨和上下颌骨,盆骨、长骨也可受累。有时开始时为单发以后多处骨组织受累,也可一开始即为多发性。骨组织被大量增生的Langerhans细胞和肉芽组织破坏。病变侵犯颅骨、硬脑膜及邻近骨组织,可累及颅底、蝶鞍及眼眶。增生的组织侵犯压迫垂体后叶和下丘脑可引起尿崩症。病变累及眼眶可压迫眼球引起眼球突出。颅骨缺损,尿崩症和眼球突出是本病的三大特征。
镜下见骨髓腔内Langerhans细胞增生,胞浆丰富,内含大量脂质,主要为胆固醇及胆固醇酯,呈泡沫状。其间有多数嗜酸性粒细胞、淋巴细胞和浆细胞浸润,常有多数多核巨细胞。病灶周围纤维组织逐渐增生,形成慢性炎性肉芽肿。病灶中央可发生坏死,有时可有出血及胆固醇结晶沉积,最后病灶纤维化形成疤痕。有些病人骨组织病灶内同时伴有大量嗜酸性粒细胸浸润,称为多发性嗜酸性肉芽肿。
皮肤病变多比Letteer-Siwe病为轻。肺、肝、脾、淋巴结等都可发生类似病变。
本病的病变呈进行性,原有病灶纤维化后,又可出现新的病灶,病程较长。病变广泛而严重者预后与Letterer-Siwe病相似,但一般预后较好,约半数可自动消退。小儿患者伴有贫血、血小板减少者预后较差,成年人预后较好。
三、嗜酸性肉芽种
嗜酸性肉芽肿是一种良性病变,多见于儿童和青年,也可见于其他年龄。男性多于女性。病变一般局限于骨骼,病灶多为单个,一般不累及皮肤或内脏。病人一般无明显症状,如病变破坏骨组织可引起疼痛。
【病理变化】
主要病变为骨组织内大量Langerhans细胞增生,细胞分化成熟,无明显异型性。其间并有大量嗜酸性粒细胞和多数淋巴细胞、浆细胞及中性粒细胞浸润。有时可见多核巨细胞,形态与炎性肉芽肿相似,故称之为嗜酸性肉芽肿。以后嗜酸性粒细胞逐渐减少,纤维组织增生,并出现大量吞噬脂类的泡沫细胞。最后大量纤维组织增生,病灶纤维化。骨组织内的病灶形成结节状肿块,呈灰红或灰黄色,部分可有出血,质软易碎,骨小梁被破坏,骨组织吸收,周围可有反应性新骨形成。
病变最常见于颅骨、面部骨骼、肋骨、脊柱、盆骨、肩胛骨和长骨,骨骼系统各部皆可受累。X线检查可见局限性骨质破坏,须与骨囊肿或骨肿瘤鉴别。
本病多数预后良好,病变可自行消退或经治疗后消退。
第三节 恶性淋巴瘤
恶性淋巴瘤(malignant lymphoma)是原发于淋巴结和淋巴结外淋巴组织的恶性肿瘤。在世界各地均不少见。在我国,恶性淋巴瘤的发病率在各种恶性肿瘤中居第十一位。但在儿童和青年中所占比例较高,是儿童最常见的恶性肿瘤之一。根据瘤细胞的特点和瘤组织的结构成分,可将恶性淋巴瘤分为何杰金病(Hodgkin disease,HD)和非何杰金淋巴瘤(non-Hodgkin lymphoma,NHL)两大类。
一、何杰金病
何杰金病是恶性淋巴瘤的一个独特类型,与其他恶性淋巴瘤不同,具有以下特点:①病变往往从一个或一组淋巴结开始,逐渐由邻近的淋巴结向远处扩散。原发于淋巴结外淋巴组织者较少。②瘤组织成分多样,但都有一种独特的瘤巨细胞即Reed-Sternberg细胞。瘤组织内并常有多数各种炎症细胞浸润。
本病在欧美各国发病率较同,是青年人中最常见的恶性肿瘤之一。在我国发病率较低,大致占全部恶性淋巴瘤的10%~20%。
【病理变化】
病变主要发生于淋巴结,以颈部淋巴结和锁骨上淋巴结最为常见,其次为纵隔、腹膜后、主动脉旁等淋巴结。病变常从一个或一组淋巴结开始,很少开始即为多发性。晚期可侵犯血管,累及脾、肝、骨髓和消化道等处。
肉眼观察,病变的淋巴结肿大,早期无粘连,可活动。瘤组织浸润淋巴结包膜,并侵入邻近组织时则不易推动。相邻的淋巴结常相互粘连,形成结节状巨大肿块。切面灰白色呈鱼肉状,可见散在的黄色小坏死灶。
镜下,淋巴结的正常结构破坏消失,由瘤组织取代。瘤组织内的细胞成分多样,有些是肿瘤成分,有些是非肿瘤成分。瘤组织中有一种独特的多核瘤巨细胞,体积大,直径约15~45μm,椭圆形或不规则形;胞浆丰富,双色性或呈嗜酸性;核大,可为双核或多核,染色质常沿核膜聚集成堆,核膜厚。核内有一大的嗜酸性核仁,直径约3~4μm,周围有一透明晕。这种细胞称为Reed-Sternberg细胞(R-S细胞)。双核的R-S细胞的两核并列,都有大的嗜酸性核仁,形似镜中之影故称镜影细胞(图11-1)。这些双核和多核的R-S细胞是诊断HD的重要依据。
图11-1 何杰金病示镜影细胞
何杰金病的肿瘤成分中除典型的R-S细胞外,还有一些肿瘤细胞,形态与R-S细胞相似,但只有一个核,内有大形核仁,称为何杰金细胞。这种细胞可能是R-S细胞的变异型,不能作为诊断的依据。此外,还有一些变异的R-S细胞常见于本病的某些特殊类型:①有些细胞体积较大,胞浆淡染,核大,常扭曲,呈折叠状或分叶状;核膜薄,染色质细,核仁小,可有多个小核仁。这种细胞常见于淋巴细胞为主型何杰金病。②陷窝细胞(lacunar cell)多见于结节硬化型何杰金病。细胞体积大,胞浆丰富,染色淡或清亮透明,核大呈分叶状,常有多个小核仁。用福尔马林固定的组织,细胞浆收缩与周围细胞之间形成透明的空隙,好似细胞位于陷窝内故称陷窝细胞(图11-2)。③多形性或未分化型细胞,多见于淋巴细胞消减型何杰金病。细胞体积大,大小形态多不规则;核大,形状不规则,核膜厚,染色质粗,常有明显的大形核仁,核分裂像多见,并常有多极核分裂。
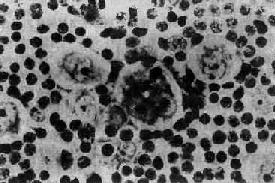
图11-2 何杰金病示陷窝细胞
除上述肿瘤细胞外,瘤组织间还有多数非肿瘤成分,包括淋巴细胞、浆细胞、中性粒细胞、嗜酸性粒细胞和组织细胞等,数量多少不等。除特征性的R-S细胞外必须同时伴有多样化的反应性非肿瘤性细胞背景,才能诊断为何杰金病。瘤组织内的非肿瘤性成分主要是淋巴细胞,可反映机体的免疫状态。淋巴细胞的多少与何杰金病的扩散和预后有密切关系。
关于R-S细胞的来源意见不一。由于有些患者常有T细胞免疫功能缺陷,因而有人认为R-S细胞可能由T细胞恶变而来。但是免疫标记和T细胞受体(TCR)及免疫球蛋白(Ig)的基因重组分析结果不一。有些R-S细胞具有T细胞标记,有些具有B细胞标记,另有些则表现单核巨噬细胞或指突状网状细胞标记。因此R-S细胞的来源尚未肯定。
【组织类型】
根据肿瘤组织内肿瘤细胞成分与非肿瘤细胞成分的不同比例,可将何杰金病分为4种组织类型。
1.淋巴细胞为主型(lymphocyte predominance type)淋巴结内有大量淋巴细胞和数量不等的组织细胞,呈弥漫性浸润或形成结节状。嗜酸性粒细胞、中性粒细胞和浆细胞数量很少,没有坏死或纤维组织增生。其间可见典型的R-S细胞,但数量很少,可见较多数有多个小核仁的变异型R-S细胞。此型HD一般只累及一个或一组淋巴结。病人一般无明显症状,预后良好。
2.混合细胞型(mixed cellularity type)此型是何杰金病中最多见的类型。病变和预后都介于淋巴细胞为主型和淋巴细胞消减型之间,由多种细胞成分混合而成。淋巴结结构消失,内有多数嗜酸性粒细胞、浆细胞、组织细胞、淋巴细胞和少量中性粒细胞浸润。其间常有多数典型的R-S细胞,部分可有小坏死灶和少量纤维组织增生,一般不形成胶原纤维束。
3.淋巴细胞消减型(lymphocyte depletion type)本型的特点为淋巴细胞数量减少而R-S细胞或其变异型多形性细胞相对较多。这种类型有两种不同的形态:①弥漫性纤维化,淋巴结内细胞少,主要由排列不规则的纤维组织和纤细的蛋白样物质替代。其间有少数R-S细胞、组织细胞和淋巴细胞,并常有坏死灶;②网织型或肉瘤型,细胞丰富,由多数高度未分化的多形性细胞组成。其间可见少数典型的R-S细胞。瘤组织内常有坏死灶。淋巴细胞消减型何杰金病多发生于年长者,进展快,是本病各型中预后最差的。
4.结节硬化型(nodular sclerosis type)此型特点为淋巴结瘤组织内有陷窝细胞和增生的纤维组织条索。淋巴结内纤维组织增生,由增厚的包膜向内伸展,形成粗细不等的胶原纤维条索,将淋巴结分隔成许多大小不等的结节。其中有多数陷窝细胞和多少不等的典型的R-S细胞。此外,还可见较多淋巴细胞、组织细胞、嗜酸性粒细胞、浆细胞和中性粒细胞浸润,部分可有坏死。
本型为一特殊类型,多见于青年妇女,是何杰金病中唯一多见于女性的类型。多发生于颈部,锁骨上和纵隔淋巴结,预后好。
上述组织类型在疾病过程中可以转化。淋巴细胞为主型可转变为混合细胞型或淋巴细胞消减型。混合细胞型可转变为淋巴细胞消减型。结节硬化型一般不转变为其他类型。
何杰金病的组织类型与预后有密切关系,一般以淋巴细胞为主型预后最好,其次为结节硬化型和混合细胞型,淋巴细胞消减型预后最差。
【何杰金病的分期】
何杰金病扩散时多由近及远。根据病变范围可分为四期。
Ⅰ期:病变限于一个淋巴结或限于一个淋巴结外器官。
Ⅱ期:病变局限于膈的一侧。单独累及2个以上的淋巴结区或同时直接蔓延至相邻的淋巴结外器官或组织。
Ⅲ期:膈两侧淋巴结都受累,可累及脾、并直接蔓延到邻近的淋巴结外器官。
Ⅳ期:肿瘤扩散至淋巴结外。累及一个或多个淋巴结外器官或组织。
何杰金病的病变范围与预后有密切关系,病变范围越广,预后越差。临床上常根据病变范围决定治疗方案。
【临床病理联系】
本病的主要表现为无痛性淋巴结肿大。早期多无明显症状,较晚期病变扩散,患者常有发热、盗汗、体重减轻、乏力、皮肤骚痒、贫血等全身症状,并常有免疫功能(主要是T细胞免疫功能)低下,容易并发感染,如疱疹病毒和隐球菌感染等。感染和肿瘤广泛扩散是招致何杰金病患者死亡的重要原因。
近年来由于诊断和治疗方法的改进,本病的预后显著改善。但是长期使用化疗和放射治疗的患者发生急性白血病和非何杰金淋巴瘤者增多。
二、非何杰金淋巴瘤
非何杰金淋巴瘤(NHL,简称淋巴瘤)多发生于表浅淋巴结,以颈部淋巴结最多见,其次为腋下和腹股沟淋巴结,并可累及纵隔、肠系膜和腹膜后等深部淋巴结。近1/3的淋巴瘤发生于淋巴结外的淋巴组织,如咽淋巴环、扁桃体、胃肠和皮肤等。病变可从一个或一组淋巴结开始,逐渐侵犯其他淋巴结,也可开始即为多发性。淋巴结和结外淋巴组织的淋巴瘤都有向其他淋巴结和全身其他组织和器官如脾、肝、骨髓等扩散的倾向。有时淋巴瘤广泛播散,瘤细胞侵入血流,全身多数淋巴结和骨髓内都可有瘤细胞浸润,很难与白血病侵犯淋巴结相区别。非何杰金淋巴瘤与何杰金病不同,瘤组织成分单一,以一种细胞类型为主。故常根据瘤细胞的类型鉴别其来源,是NHL分类的基础。
【分类】
非何杰金淋巴瘤的分类方法很多,但大多仍以Lukes-Collins淋巴瘤的免疫功能分类为基础。因此本章重点介绍Lukes-Collins分类及其分类依据。Lukes等将近代免疫学的观念和新技术应用于淋巴瘤的研究,提出了形态与功能结合以瘤细胞来源为基础的免疫功能分类,并将本瘤分为B细胞、T细胞和组织细胞型三大类及不同的亚型(表11-1)。其中B细胞淋巴瘤最多见,T细胞淋巴瘤次之,组织细胞淋巴瘤很少见。恶性淋巴瘤是免疫系统的肿瘤。淋巴细胞在分化、成熟和转化过程中的任何阶段都可能发生恶变,形成肿瘤。淋巴细胞发育过程不同时期发生的肿瘤,在形态和免疫功能方面都与其相应的正常细胞相似。因此,了解这些正常细胞发育分化过程中的形态和功能变化,有助于理解各型淋巴瘤的发生及其特点。
表11-1 非何杰金淋巴瘤Lukes-Collins分类
| B细胞型淋巴瘤 | T细胞型淋巴瘤 |
| 小淋巴细胞(B)型 | 小淋巴细胞(T)型 |
| 浆细胞样淋巴细胞型 | 曲折核淋巴细胞型 |
| 滤泡中心细胞型 | 脑迴状核淋巴细胞型(蕈样霉菌病,Sézary综合征) |
| 小核裂滤泡中心细胞型 | 免疫母细胞型淋巴瘤(T) |
| 大核裂滤泡中心细胞型 | 组织细胞型淋巴型 |
| 小无核裂滤泡中心细胞型 | 未定型淋巴瘤 |
| Burkitt淋巴瘤 | |
| 非Burkitt淋巴瘤 | |
| 大无核裂滤泡中心细胞型 | |
| 免疫母细胞型淋巴瘤(B) |
淋巴细胞和髓样细胞在发育分化、成熟以及接受抗原刺激后转化的过程中,细胞内和细胞表面的分子结构发生一系列变化。这些细胞在发育分化各不同时期的标记可用细胞化学,免疫组织化学,单克隆抗体技术,流式细胞仪技术及分子生物学技术等加以识别。例如B细胞具有表面免疫球蛋白(SIg)。前B细胞表面无SIg,但具有CD19(B4)和CD20(B1),这些是幼稚的B细胞最早出现的表面抗原。同时前B细胞浆内先后出现μ重链基因重组和Ig轻链重组,可用分子生物学方法,如多聚酶链反应技术检测。T细胞具有羊红细胞受体(CD2)可与羊红细胞形成E花环。T细胞分化的各个阶段表现不同的表面抗原都可用特异性的单克隆抗体加以识别。此外,应用分子生物学技术分析Ig及TCR基因对鉴别B细胞和T细胞及其克隆性提供了更为精确的方法。组织细胞除表面抗原外还含有多种酶类如非特异性酯酶,α1-抗胰蛋白酶和溶菌酶等,可用组织化学和免疫组织化学方法显示。成熟淋巴细胞在受抗原刺激后,转化为免疫母细胞和产生抗体的浆细胞的过程中,形态和功能也发生一系列变化。Lukes等提出B细胞的转化过程在淋巴结的淋巴滤泡生发中心进行,大致可分为4个时期:①小核裂细胞,②大核裂细胞,③小无核裂细胞和④大无核裂细胞(图11-3)。成熟的B细胞接受抗原信息后体积逐渐增大,细胞核表面出现沟状凹陷似裂隙称为小核裂细胞。细胞继续增大,核较大,周围有少量胞浆,称为大核裂细胞。以后核逐渐变为圆形或椭圆形,裂隙消失,核内出现核仁。胸浆增多,胞浆内RNA含量和蛋白质合成增多,派若宁染色阳性,称为小无核裂细胞。这时滤泡中心内可见多数核分裂像。细胞继续增大可达原来小淋巴细胞的4~6倍。核大,圆或椭圆形,核内常有1~3个明显的核仁。胞浆丰富,派若宁染色阳性,称为大无核裂细胞,核分裂像多见。以上4种细胞都属于滤泡中心细胞(follicular center cell)。大无核裂细胞逐渐向淋巴滤泡外移动,进入滤泡间区,继续增大形成免疫母细胞,体积大,胞浆丰富,派若宁染色强阳性,核大,圆或椭圆形,有明显的核仁,有些可表现浆细胞样的特点。免疫母细胞继续增殖形成浆细胞或转变为记忆细胞。应用免疫细胞化学和细胞表面标记检查证实,滤泡中心细胞属于B细胞。
图11-3 淋巴细胞转化过程示意图
T细胞分布于淋巴结的副皮质区,受抗原刺激后也发生相应的转化过程,形成T免疫母细胞。T细胞在转化过程中与B细胞不同,没有核裂细胞到无裂细胞的各种变化。有些T细胞来源的淋巴瘤,瘤细胞胞浆少,核形状不规则,呈多角形,或有裂隙呈折叠状或分叶状,称为曲折核淋巴瘤。瘤细胞含有末端去氧核苷酸转移酶,免疫表型为CD2+,CD7+,CD5+,这些都是正常未成熟T细胞具有的标记,因此,说明瘤细胞来源于未成熟T细胞。
各型非何杰金淋巴瘤细胞的形态和免疫标记在不同程度上与其来源的相应的正常细胞相似,故对其形态不另做详细描述。病变淋巴结内有大量瘤细胞浸润将淋巴结的结构破坏。瘤细胞成分单一,以一种细胞类型为主,其特点与其来源的细胞相似,但具有一定程度的异型性。可参阅正常淋巴细胞的形态及其分化抗原或表面标记来鉴别其类型。
除Lukes-Collins的免疫功能分类外,还有Rappaport于1966年提出的分类,主要根据常规组织切片中淋巴瘤细胞的形态和组织结构决定淋巴瘤的类型,曾广泛应用。因提出的年代较早,其中有些名称不符合现代科学的观点。但由于Rappaport分类方法简便易行,有些地方仍沿用至今。此外,还有很多分类方法,对临床应用造成了困难和混乱。因此美国国家癌症研究所组织了国际上著名的专家共同讨论研究制订了一个NHL的工作分类(表11-2)。这个工作分类不代表各家的统一意见,也不取代原有的分类。工作分类的作用在于将各种分类法中相应的类别和名称联系起来,以便在具体工作中有共同的了解。工作分类中所应用的名称比较混杂,多数与Lukes-Collins分类相似。
表11-2 非何杰金淋巴瘤的工作分类及其与Lukes-Collins分类和Rappaport分类的关系
| 工作分类 | Lukes-Collins分类 | Rappaport分类 |
| 低度恶性 | ||
| A.小淋巴细胞型 | 小淋巴细胞和浆细胞样淋巴细胞型 | 高分化淋巴细胞性 |
| B.滤泡性,小核裂细胞为主型 | 滤泡中心细胞,小核裂细胞型 | 结节性低分化淋巴细胞性 |
| C.滤泡性,小核裂和大核裂细胞混合型 | 滤泡中心细胞,小核裂和大核裂细胞混合型 | 结节性,淋巴细胞-组织细胞混合性 |
| 中度恶性 | ||
| D.滤泡性,大细胞为主型 | 滤泡中心细胞,大核裂/无核裂细胞型 | 结节性,组织细胞性 |
| E.弥漫性,小核裂细胞型 | 滤泡中心细胞,弥漫性小核裂细胞型 | 弥漫性,低分化淋巴细胞性 |
| F.弥漫性,大、小细胞混合型 | 滤泡中心细胞,小核裂细胞,大核裂细胞或大无核裂细胞型 | 弥漫性,淋巴细胞-组织细胞混合性 |
| G.弥漫性,大细胞型 | 滤泡中心细胞,大核裂细胞或大无核裂细胞型 | 弥漫性,组织细胞性 |
| 高度恶性 | ||
| H.大细胞,免疫母细胞型 | 免疫母细胞型,B或T细胞性 | 弥漫性,组织细胞性 |
| I.淋巴母细胞型 | 曲折核T细胞型淋巴瘤 | 淋巴母细胞性淋巴瘤 |
| J.小无核裂细胞型 | 滤泡中心细胞,小无核裂细胞型 | 未分化型,Burkitt和非Burkitt |
| 杂类 | ||
| 复合性 | ||
| 蕈样霉菌病 | 脑回状T细胞淋巴瘤(蕈样霉菌病) | |
| 组织细胞型 | 组织细胞型 | |
| 髓外性浆细胞瘤 | ||
| 不能分类者 | ||
| 其他 |
【临床病理联系】
多数患者起病缓慢,早期多无症状。主要表现为无痛性淋巴结肿大。晚期病变可累及多处淋巴结或其他器官。根据受累的器官不同可引起不同的症状。NHL的扩散途径与HD不同,多无一定规律。晚期病人常有发热、盗汗、消瘦及肝、脾肿大。
儿童淋巴瘤与成人淋巴瘤有些不同,淋巴结外器官的淋巴瘤比较多见。曲折核T细胞淋巴瘤伴纵隔肿块者和Burkitt淋巴瘤常见于儿童,前者常伴有急性淋巴细胞性白血病,预后很差。NHL的预后与病变范围和肿瘤的组织类型有关。病变局限在一个部位者预后较好。多组淋巴结受累、肝脾肿大或侵犯其他器官者预后较差。肿瘤的组织类型中,一般以小淋巴细胞型、浆细胞样淋巴细胞型和有核裂的滤泡中心细胞型,尤其是早期瘤细胞呈滤泡样增生者比弥漫增生者预后较好。无核裂细胞型预后较差。免疫母细胞型和曲折核T细胞淋巴瘤预后最差。
【特殊类型的淋巴瘤】
1.Burkitt淋巴瘤 Burkitt淋巴瘤是1958年Burkitt首先描述的发生于非洲儿童的一种淋巴瘤。现在世界各地都发现类似的病例。我国也有少数病例报道。患者主要为儿童和青年人。男性多于女性。
病变特点为肿瘤常发生于颌骨、颅面骨、腹腔器官和中枢神经系统等。一般不累及外周淋巴结和脾,也很少发生白血病。颌骨和眼眶的肿瘤在局部生长,侵蚀破坏附近组织,造成面部畸形。肿瘤发生于腹腔,常形成巨大肿块,并可累及腹膜后淋巴结、卵巢、肾、肝、肠等。累及中枢神经系统的肿瘤可侵犯脑膜或压迫脊髓。
肿瘤由小无核裂滤泡中心细胞恶性增生而来。镜下见大量瘤细胞弥漫增生,细胞大小相似,形态单一,胞浆少,呈嗜碱性及明显的嗜派若宁性。胞浆内有一些脂肪小空泡。细胞核较大,圆或椭圆形,染色质细,常有2~3个明显的核仁,核分裂像多见。肿瘤细胞常变性、坏死。瘤细胞表面有单克隆性免疫球蛋白,多数为IgM伴κ轻链,证实瘤细胞来自B细胞。瘤细胞间散在多数吞噬各种细胸碎屑的巨噬细胞,形成所谓满天星图像(图11-4)。
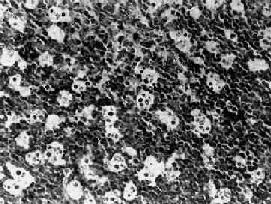
图11-4 Burkitt淋巴瘤
瘤细胞由小无核裂滤泡中心细胞组成,细胞大小相似,核圆形。瘤细胞间散在多数吞噬细胞呈“满天星”图像
Burkitt淋巴瘤对化疗效果较好,缓解期长。本病原因尚不清楚,由于常发生在温暖潮湿地带,故有人认为可能与蚊子或其他昆虫传播的病毒性感染有关。非洲流行区EB病毒感染率很高。约95%非洲Burkitt淋巴瘤细胞有EB病毒基因组,并且Burkitt淋巴瘤患者抗EB病毒抗体滴度增高。因此有人认为EB病毒与Burkitt淋巴瘤的发生有关。
此外,还有一组淋巴瘤也来源于小无核裂细胞,并具有与Burkitt淋巴瘤相似的细胞标记,但瘤组织结构和病人的预后与Burkitt淋巴瘤不同,称为非Burkitt淋巴瘤。非Burkitt淋巴瘤的瘤细胞比较多形性,大小不一,细胞核大小也不一致。核仁大而明显,有时只有一个大核仁。巨噬细胞造成的“满天星”现象不多见(图11-5)。非Burkitt淋巴瘤比较少见。多发生于成年人,治疗效果较差,预后也较差。
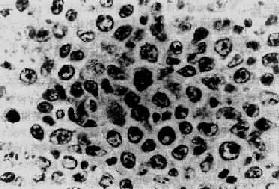
图11-5 小无核裂滤泡中心细胞型淋巴瘤—非Burkitt淋巴瘤
瘤细胞大不不甚一致,胞浆少,核多为圆形,大小形状也不一致
2.蕈样霉菌病 蕈样霉菌病(mycosis fungoides)是原发于皮肤的T细胞淋巴瘤。多发生于40~60岁。男性多于女性,约为2:1。
肉眼观,皮肤病变早期表现为湿疹样病损,皮肤瘙痒,表面有不规则的红色或棕色斑疹,逐渐发展使皮肤增厚变硬呈斑块状。以后形成多数不规则的棕红色瘤样结节。有时可溃破。
镜下可见皮肤表皮和真皮浅层及血管周围有多数瘤细胞和嗜酸性粒细胞、淋巴细胞、浆细胞和组织细胞浸润。瘤细胞体积较大,核大,染色深,高度扭曲,有深切迹,呈折叠状或脑回状。这些瘤细胞SIg阴性,与羊红细胞形成E花环,CD4+,来自T辅助细胞。真皮内的瘤细胞常侵入表皮,在表皮内聚集成堆形似小脓肿称为Pautrier微脓肿。晚期表皮可溃破,并可向深部浸润及真皮深部。
蕈样霉菌病早期病变局限于皮肤,以后常扩散至淋巴结和内脏,多见于脾、肝、肺和骨髓等处。病变局限于皮肤者治疗效果较好,扩散至内脏者预后较差。
第四节 白血病
白血病(leukemia)是一种造血干细胞的恶性肿瘤。其特征为骨髓内异常的白细胞(白血病细胞)弥漫性增生取代正常骨髓组织,并常侵入周围血液,使周围血内白细胞出现量和质的改变。血液白细胞数量常明显增多,但有时亦可正常甚至减少。白血病细胞并可广泛浸润肝、脾、淋巴结等全身各组织和器官,并常导致贫血和出血。
白血病在我国和世界各地都不少见。在我国各种恶性肿瘤死亡率中居第六或第七位。但在儿童和青少年的恶性肿瘤中,白血病则居第一位。
【分类】
白血病有几种分类方法。
1.成熟程度可分为急性与慢性白血症。急性白血病起病急,病程短,骨髓和周围血中以异常的原始及早期幼稚细胞为主,原始细胞常超过30%。慢性白血病病程缓慢,骨髓及周围血中以异常成熟的白细胞为主,伴有幼稚细胞。原始细胞一般不超过10%~15%。
2.根据增生细胞的类型可分为淋巴细胞性和粒细胞性白血病两类。结合病情急缓和细胞类型,白血病总的可分为4种基本类型:①急性淋巴细胞性白血病(acute lymphocytic leukemia,ALL),②慢性淋巴细胞性白血病(chronic lymphocytic leukemia,CLL),③急性粒细胞性白血病(acute myelocytic leukemia,AML)或急性非淋巴细胞性白血病(acute non-lymphocytic leukemia,ANLL),④慢性粒细胞性白血病(chronic myelocytic leukemia,CML)。
急性白血病与慢性白血病的临床表现、血像、骨髓像以及治疗原则都不相同。急性白血病经过治疗后可进入缓解期,一般不转变为慢性。慢性白血病后期常有急性变,临床表现很象急性白血病。
3.根据周围血内白细胞的数量可分为白细胞增多性白血病(周围血内白细胞数量增多,>15000/μ1)和白细胞不增多性白血病(周围血内白细胞数量不增多,甚至减少)。
4.白血病的免疫学分类应用单克隆抗体和分子生物学技术检测白血病细胞的免疫标记可鉴别白血病细胞的来源,如急性淋巴细胞性白血病可分为T细胞型,B细胞型,前-B细胞型和无标记细胞型等。
在我国大多数地区,急性白血病比慢性白血病多见。无论急性或慢性白血病均以粒细胞性占多数,急性淋巴细胞性白血病次之,慢性淋巴细胞性白血病最少见。急性白血病多见于儿童和青少年,慢性粒细胞性白血病多见于30~50岁。慢性淋巴细胞性白血病多发生在50岁以上。各种类型的白血病都较多见于男性。
一、急性白血病
急性白血病起病急,常表现为发热、乏力、进行性贫血、出血倾向、淋巴结及肝、脾肿大等。
急性白血病时造血干细胞或原始和幼稚的白细胞恶变,发生分化障碍,不能分化为成熟的细胞,使骨髓内原始和幼稚细胞大量堆积,成熟的细胞明显减少。大量异常的原始和幼稚幼胞增生,抑制正常的造血干细胞和血细胞生成,引起红细胞、白细胞和血小板减少。多能造血干细胞分化过程中的任何阶段都可能发生恶变转化为白血病细胞。根据累及的细胞类型可分为急性淋巴细胞性白血病(ALL)和急性粒细胞性白血病(AML)。
(一)急性淋巴细胞性白血病
本病多见于儿童和青年人。根据形态学和免疫学特点可分为不同的亚型。
1.国际上通用的法、美、英协作组的FAB分类,根据瘤细胞形态将急性淋巴细胞性白血病分为L1,L2,和L3型。
L1型:细胞较小,大小一致。核较大,圆形,染色质均匀、细致,核仁不明显。胞浆少,嗜碱性。这种类型多见于儿童。
L2型:细胞较大,约为正常成熟小淋巴细胞的2倍。细胞大小不一,胞浆丰富,嗜碱性。细胞核形状不规则,有些有裂隙或切迹或呈折叠状,常有1~2个明显的核仁。这种类型多见于成人或大年龄儿童。
L3型:细胞大,大小一致。胞浆丰富,嗜碱性,胞浆内常有小空泡。细胞核圆或椭圆形,外形规则。染色质致密、均匀,呈点彩状,常有一个或多个明显的核仁。
2.免疫学分类 应用免疫标记和TCR及Ig基因重组技术,根据白血病细胞的来源和分化的不同阶段分类。免疫学分类与预后有关,有助于指导临床治疗。
(1)B细胞性ALL(B-ALL):ALL中约80%来源于B细胞。可分为3种亚型,其中2种来自原始B细胞。①前B细胞ALL,约占ALL的60%。来自早期的原始B细胞,CD19+,CD10+,预后最好;②前B细胞ALL,约占ALL的20%,CD19+,CD10+,CD20+,Cμ+,预后较好;③较成熟的B细胞ALL很少见,约占ALL的1%~2%,除CD19+,CD20+外SIg+,预后最差。
(2)T细胞性ALL(T-ALL):约占ALL的15%,来自原始T细胞,CD2+,CD7+,CD5+,预后较差。
(3)无标记(未分类)ALL:在ALL中<5%,没有B细胞和T细胞标记,预后较差。
(二)急性粒细胞性白血病
多见于成人,儿童较少。多能髓细胞样干细胞在分化过程的不同阶段都可发生恶变,因此AML的细胞来源不同,可分为多种类型。FAB分类根据白血病细胞分化的程度和主要的细胞类型分为M1至M7七个类型。
M1 急性原粒细胞白血病未分化型大多数瘤细胞为原粒细胞,少数为早幼粒细胞。
M2 急性原粒细胞白血病分化型瘤细胞包括多数原始粒细胞和多数早幼粒细胞及多少不一的中幼粒以下的细胞。
M3 急性早幼粒细胞白血病以早幼粒细胞为主,胞浆内充满髓过氧化物酶阳性颗粒。
M4 急性粒-单核细胞白血病瘤细胞包括粒细胞及单核细胞两种方向分化。粒细胞同M2,但同时有多数幼单核细胞和单核细胞。
M5 急性单核细胞白血病以原单核细胞为主或以幼单核细胞为主。
M6 急性红白血病瘤细胞以畸形、多核或分叶状核的原红细胞为主,同时有原单核细胞和早幼单核细胞。
M7 急性巨核细胞白血病主要为多形性、未分化的原巨核细胞。
【病理变化】
白血病的特点是骨髓内异常白细胞大量增生,进入周围血并可浸润肝、脾、淋巴结等全身各组织和器官。增生的白血病细胞形态与其来源的相应正常细胞相似,但分化不成熟,有一定的异型性。各种白血病类型虽然不同,但引起的病变有许多共同之处,包括大量白血病细胞增生直接引起的病变和白血病细胞浸润各组织、器官引起的继发性病变。
1.周围血像早期即出现贫血,白细胞总数多少不等,白细胞增多性与不增多性者约各占一半。白细胞增多性者,白细胞总数多在2万~5万/μl,常呈进行性上升,可高达10至数10万。其中有大量原始和幼稚细胞(图11-6)。白细胞不增多性白血病的白细胞计数可正常或减少,有时可降低至1000~3000/μ1,较难找到原始或幼稚细胞。血小板减少有时达1万/μ1以下。
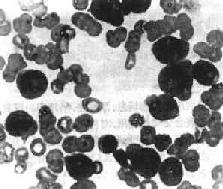
图11-6 急性粒细胞性白血病
周围血内有大量原始粒细胞
2.骨髓骨髓内白血病细胞大量增生,可取代正常骨髓组织,并可侵蚀骨松质和骨皮质。病变以椎骨、胸骨、肋骨和盆骨最显著,严重者可侵犯长骨。急性白血病的骨髓增生极为活跃,其中主要为原始细胞,较成熟的白细胞不多,幼稚红细胞和巨核细胞生成受抑制,数量减少。有些急性粒细胞白血病时大量瘤细胞主要为原粒细胞,在骨组织、骨膜下或软组织中浸润,可聚集形成肿块,称为绿色瘤(chloroma),多见于颅骨和眼眶周围。瘤细胞浸润之处呈绿色,暴露于空气中后,绿色迅速消退。用还原剂(过氧化氢或亚硫酸钠)可使绿色重现。绿色色素的性质还不肯定,有人认为其中含有原卟啉、胆绿蛋白或绿色过氧化物酶,可能与瘤细胞的异常代谢产物有关。
3.淋巴结 全身淋巴结都可有不同程度的肿大,以儿童ALL时较多见,也最明显,AML时较轻。肿大的淋巴结一般不互相粘连,有弹性。切面呈均匀的灰白色。镜下可见淋巴结内有大量瘤细胞浸润。淋巴结结构可部分或全部被破坏。瘤细胞可侵犯淋巴结包膜及包膜外脂肪组织。AML时淋巴结内瘤细胞浸润较少,部分淋巴结结构可保留。
4.脾 急性白血病时脾轻度至中度肿大。儿童ALL时脾肿大较多见,也较明显。成人AML时多轻度肿大。肉眼观,肿大的脾包膜紧张,呈暗红色,质软。镜下,ALL时红髓和血窦内有大量白血病细胞浸润,可形成结节状。脾小体可增大或消失。AML时,主要累及红髓,原粒细胞增生,可压迫脾小体,严重时红髓和脾小体结构可被破坏。
5.肝 肝中度增大,表面光滑。镜下,AML时瘤细胞主要沿肝窦在肝小叶内弥漫浸润。ALL时瘤细胞主要浸润于汇管区及其周围的肝窦内。单核细胞性白血病较少累及肝,其浸润方式与粒细胞白血病相似。
除上述器官外,急性白血病时白血病细胞还常浸润脑、脊髓、周围神经、心肌、肾、肾上腺、甲状腺、睾丸和皮肤等乃至全身各器官和组织。瘤细胞多首先出现在血管周围,逐渐向邻近组织浸润,可引起出血并可压迫和破坏邻近组织。
白血病的皮肤病多见于急性单核细胞白血病(M5)。病变多样,可局限于一处也可播散到身体大部,形成各种扁平或隆起的斑块或丘疹。瘤细胞多浸润于真皮内,一般不侵犯表皮。有时单核细胞白血病可浸润牙龈,使牙龈粘膜肿胀肥厚,常有出血,并可形成表浅溃疡,易引起继发感染。
6.继发性变化由于白血病细胞广泛浸润,常破坏相应的组织或器官,引起一系列继发性改变:
(1)出血:白血病细胞浸润骨髓组织,引起贫血和血小板减少,故常易出血。皮肤可有出血点和瘀斑。牙龈、肾盂、肾盏和膀胱粘膜、浆膜都可有出血灶,有时脑组织出血可形成血肿。
(2)感染:白血病时虽然白细胞大量增生但无抗病功能。患者免疫功能和抵抗力低下,常并发细菌和真菌感染,常见的有白色念珠菌、曲菌和毛霉菌感染等,成为白血病常见的致死原因。
急性白血病病情急,预后差,早期即出现贫血、出血和继发性感染,死亡率很高。近年来由于联合化疗的应用,对提高急性白血病的缓解率,延长生存期都取得了良好的效果。尤其是儿童ALL可长期缓解。
二、慢性白血病
慢性白血病起病缓慢,病程长,早期多无明显症状。有些病人在体格检查或因其他疾病就诊时发现。主要症状为乏力、消瘦、发热、脾肿大等。
慢性白血病按细胞来源分为慢性粒细胞白血病(CML)和慢性淋巴细胞白血病(CLL)。
CML来源于多能髓样干细胞。瘤细胞的成分以幼粒细胞为主。骨髓和脾内幼稚粒细胞明显增多。与急性白血病不同,CML时髓样干细胞仍具有分化成熟的能力,周围血内可见大量成熟的粒细胞。原始粒细胞的异常增生与分化,可能与干细胞对调节造血细胞生长分化的反馈信号反应不足有关。
CML病人中约90%伴有一种染色体异常,称为Ph1染色体,已确定为CML的标记染色体。Ph1染色体指22号染色体的长臂易位至9号染色体长臂。Ph1染色体的存在与CML的发生发展有关。典型的CML时Ph1阳性,多见于青壮年,化疗效果好。Ph1阴性的CML多见于老人和小儿,预后不佳。
CLL为小淋巴细胞恶变而来。绝大多数CLL来源于B细胞,T细胞性CLL很少见。恶变的B细胞无免疫功能,不能转化为浆细胞,病人常有低丙种球蛋白血病。有时CLL与小淋巴细胞淋巴瘤不易区别。
【病理变化】
1.周围血像周围血白细胞显著增多,数量正常或减少者少见。CML时白细胞增多尤为明显,多在10万~至80万/μ1之间,少数甚至可超过100万,其中大多数为较成熟的中、晚幼和杆状核粒细胞,早幼粒和原粒细胞很少(图11-7)。嗜碱性和嗜酸性粒细胞也增多。CML时中性粒细胞内碱性磷酸酶常缺如或降低。这点有助于与类白血病反应相区别。CLL血白细胞总数多在3万~10万/μ1之间。血像单一,绝大多数为成熟的小淋巴细胞,只有少数幼淋巴细胞。慢性白血病早期贫血较轻,血小板无明显减少。CML时血小板常增多。晚期有明显贫血和血小板减少。
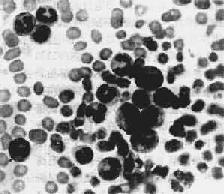
图11-7 慢性粒细胞性白血病
周围血内白血病细胞主要为中幼粒细胞
2.骨髓 CML时骨髓增生活跃,各期粒细胞均可见到,以中、晚幼粒和杆状粒细胞占优势,幼红细胞和巨核细胞早期可增生,血小板增多,晚期则被抑制。CLL骨髓内淋巴细胞增多,可呈结节状或弥漫性浸润,主要为成熟的小淋巴细胞,原及幼淋巴细胞很少。粒、红、巨核细胞系及血小板均减少。
3.淋巴结 CLL时淋巴结明显肿大。早期淋巴结可活动,晚期瘤细胞浸润包膜,肿大的淋巴结常相互融合,并与周围组织粘连。切面灰白色鱼肉状。镜下可见淋巴结内有大量瘤细胞浸润,严重者淋巴结构破坏消失。CML时淋巴结轻至中度肿大,不如CLL明显。
4.脾 脾多明显肿大,CML最显著,可达4000~5000g。肿大的脾占据腹腔大部,甚至可达盆腔。红髓脾窦内有大量白血病细胞浸润,可压迫血管引起梗死。CLL时,脾肿大不如CML时严重,一般不超过2500g。肿大的脾包膜增厚,常有纤维性粘连,质较硬,切面暗红色,脾小体不明显,呈均质状。镜下见大量白血病细胞浸润,CLL时主要累及白髓,严重者白血病细胞弥漫浸润,脾小体和脾髓结构消失。
5.肝肝中度肿大,表面光滑。镜下,各型白血病的浸润方式不同。CLL时,瘤细胞多浸润于汇管区及其周围;CML时,瘤细胞多沿肝窦呈弥漫性浸润。
此外,慢性白血病时,白血病细胞并常浸润胃肠、心、肾、皮肤等全身器官和组织。白血病细胞浸润处可破坏相应的组织和器官而引起出血、感染等继发性变化。
慢性白血病病变发展缓慢,病程较长。开始约2~3年病情稳定,对化疗有效,有时稳定期可达10年以上。以后治疗无效,病情加重。CML最后常发生急性变,突然出现原因不明的高热,脾迅速肿大,贫血、血小板减少,出血症状加剧,骨及关节疼痛,骨髓和血中原粒和早幼粒细胞突然增加。急性变发生后病情常急转直下,预后很差。CLL发生急性变者极少。CLL患者平均寿命比CML长,最后多因低丙种球蛋白血症,免疫功能低下并发感染而致死。
【类白血病反应】
类白血病反应(leukemoid reaction)通常是由于严重感染、某些恶性肿瘤、药物中毒、大量出血和溶血反应等刺激造血组织而产生的异常反应,表现为周围血中白细胞显著增多(可达50,000/μ1以上),并有幼稚细胞出现。类白血病反应的治疗和预后均与白血病不同。一般根据病史、临床表现和细胞形态可以与白血病鉴别,但有时比较困难。类白血病反应有以下特点可协助鉴别:①引起类白血病反应的原因去除后,血像可恢复正常;②类白血病反应时,一般无明显贫血和血小板减少;③类白血病反应时,粒细胞有严重毒性改变,胞浆内有毒性颗粒和空泡等;④类白血病反应时,中性粒细胞的碱性磷酸酶活性和糖皆明显增高,而粒细胞白血病时,两者均显著降低;⑤慢性粒细胞白血病细胞内可见Ph1染色体,类白血病反应时则无。
三、毛细胞白血病
毛细胞白血病(hairy cell leukemia)是一种少见的慢性白血病,其主要特点为白血病细胞胞浆形成细长的突起,形似绒毛,在扫描电镜(图11-8)、透射电镜或相差显微镜下清晰可见,在一般光学显微镜下也可见到,故称毛细胞。关于毛细胞的来源意见不一。过去曾认为毛细胞来源于单核巨噬细胞。细胞标记研究证明绝大多数毛细胞具有B淋巴细胞标记,少数具有T细胞标记。毛细胞胞浆内含有抗酒石酸的酸性磷酸酶。这点与单核细胞和T细胞不同可供鉴别。应用Ig基因重组技术证实毛细胞来源于B细胞系。近来有学者提出毛细胞具有B细胞的特点,也有一些特点与单核吞噬细胞系统的树突状细胞(dendritic cells)相似,因而提出毛细胞可能由多能造血干细胞恶性增生而来,故具有B细胞和树突状细胞两者的特点。
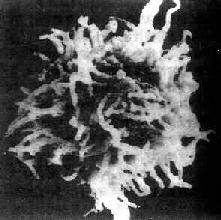
图11-8 毛细胞性白血病细胞
瘤细胞表面有多数细长的微绒毛(扫描电镜)
毛细胞白血病主要累及骨髓、脾和周围血液。骨髓内瘤细胞弥漫性增生,细胞间网状纤维弥漫增生。骨髓内髓细胞系减少可引起周围血粒细胞和单核细胞减少。脾内常有大量瘤细胞弥漫浸润。脾肿大是最常见的症状,并常引起全血细胞减少。瘤细胞常侵入周围血。大多数病人周围血及骨髓中可检见毛细胞。病人周围血白细胞数量可增高或减少。少数病例瘤细胞可浸润至肝和淋巴结。此外,偶尔瘤细胞可侵犯皮肤、肺及体腔。
毛细胞白血病平均发病年龄约为50岁,多见于男性。起病缓慢,主要表现为贫血,粒细胞、单核细胞及血小板减少和脾肿大。常呈慢性经过。治疗效果不理想。近来应用α-干扰素治疗效果良好。由于贫血、白细胞减少和免疫功能低下,晚期易并发感染。
第五节 骨髓增生性疾病
骨髓增生性疾病(myeloproliferative disorders)是多能髓样干细胞肿瘤性增生引起的一组疾病,常表现为多能髓样干细胞所属的细胞系(包括红细胞、血小板、粒-单核细胞系)中的一系或多系细胞恶性增生。根据增生的细胞成分不同,本组疾病可分为慢性粒细胞白血病,真性红细胞增多症,原发性血小板增多症和骨髓纤维化4种疾病。这4种疾病虽表现各不相同,但可相互转化(慢性粒细胞白血病已如前述)。
一、真性红细胞增多症
真性红细胞增多症(polycythemia vera)是多能髓样干细胞恶性增生引起的疾病。骨髓内常同时有红细胞、粒细胞和巨核细胞系各种成分增生,但以红细胞系增生最突出,导致红细胞明显增多,可达6~10×106/μ1。由于血红细胞增多引起血容量增多,血粘度增高,全身组织和器官淤血,血流缓慢。常有血栓形成和梗死,多见于心、脾、肾。血管严重充血和血小板功能异常,常引起出血。肝、脾轻至中度肿大,可出现髓外造血灶。血栓形成和出血是造成部分病人死亡的原因。晚期有些病人可转变为骨髓纤维化或急性粒细胞白血病。
二、原发性血小板增多症
原发性血小板增多症(primary thrombocythemia)少见,是骨髓增生性疾病中以巨核细胞系过度增生为主的疾病。血小板数量明显增多,功能异常。多发生于中年人,主要表现为反复出血和血栓形成。骨髓内巨核细胞系显着增生。脾、肝及淋巴结内可见髓样化生灶,其中的细胞以巨核细胞系为主。部分病例可转化为真性红细胞增多症、骨髓纤维化或慢性粒细胞白血病,最终可转变为急性粒细胞白血病。重要器官血栓形成及出血为主要致死原因。
三、骨髓纤维化
骨髓纤维化(myelofibrosis)是一种慢性骨髓增生性疾病。骨髓造血组织由纤维组织取代,脾、肝、淋巴结等有增生的髓样干细胞,故本病常称为骨髓纤维化伴髓样化生。骨髓纤维化可继发于其他骨髓增生性疾病、骨转移性癌、化学药物中毒和射线损伤等。多数骨髓纤维化原因不明,称为原发性骨髓纤维化。
原发性骨髓纤维化是一种多能造血干细胞恶性增生性疾病。红、粒及巨核细胞系均呈恶性增生。病变由骨髓开始,以后侵入周围血,逐步侵犯脾、肝等其他器官,并在其中增殖,形成髓样化生灶。骨髓内增生的巨核细胞和血小板释放的生长因子(PDGF和TGF-β)可刺激纤维母细胞增生形成胶原纤维,引起骨髓纤维化。血小板释放的血小板因子4可抑制胶原酶,对骨髓纤维化可能也起一定作用。
主要病变为髓外造血。最常见于脾,多分布在红髓,其中可见红、粒和巨核细胞系各期的细胞增生,并常有不同程度的异型性。脾明显肿大可达4000g,包膜下可见梗死灶。肝和淋巴结内也可有髓外造血。早期骨髓内髓样干细胞增生,网状纤维增多,以后形成胶原纤维,引起骨髓弥漫纤维化,细胞少,造血组织由增生的纤维组织取代。晚期可有新骨形成,骨小梁增粗引起骨硬化。
骨髓纤维化多发生于50岁以上的病人。病变进展慢,早期无症状,症状出现时骨髓已纤维化。主要表现为进行性贫血和脾肿大,周围血红细胞大小、形状不一,有泪滴状红细胞,并有不成熟的红细胞和白细胞。由于血小板异常和血小板减少,容易发生血栓及出血,并常并发感染威胁生命。约10%的病人转变为急性粒细胞白血病。
第六节 多发性骨髓瘤
多发性骨髓瘤(multiple myeloma)是浆细胞的恶性肿瘤。骨髓内肿瘤性浆细胞增生,常侵犯多处骨组织,引起多发性溶骨性病损。肿瘤性浆细胞可合成并分泌免疫球蛋白。由于肿瘤性浆细胞为单克隆性,故所产生的免疫球蛋白为均一的、类型相同的单克隆免疫球蛋白,具有相同的重链和轻链。浆细胞除合成完全的免疫球蛋白外,也可合成过多的轻链或重链。多发性骨髓瘤时血液内的这种单克隆Ig称为M蛋白或M成分。有时肿瘤性浆细胞只合成轻链或重链而没有完整的Ig。这种游离的轻链称为Bence-Jones蛋白,分子小,可通过肾由尿排出。99%的多发性骨髓瘤病人血液内都有一种免疫球蛋白增高。病人的M成分多数为IgG和IgA,少数为IgM、IgD或IgE。约15%~20%病人尿中有Bence-Jones蛋白,但血中无M成分。约80%的骨髓瘤病人血中M成分和尿液中Bence-Jones蛋白两者都有。这是诊断多发性骨髓瘤的重要指标。本病发病年龄多在50~60岁之间,40岁以前少见。男性多于女性。
【病理变化】
最突出的病变为骨髓内大量浆细胞增生,可占骨髓内细胞总数的15%~90%。瘤细胞多聚集成堆,有些像成熟的浆细胞,有些分化不成熟,具有不同程度的异型性。有些细胞体积大,有2或3个核,并有瘤巨细胞形成。电镜下可见骨髓瘤细胞胞浆内有高度发达的粗面内质网,其池内常充满无定形物质(免疫球蛋白)。
多发性骨髓瘤病变为多发性,常引起多处骨组织破坏,可累及骨骼系统的任何部位,以脊柱、肋骨、颅骨最多见,盆骨、股骨、锁骨和肩胛骨次之。瘤组织在骨髓腔内形成灰红色结节。瘤细胞产生破骨细胞活化因子,激活破骨细胞使骨质溶解吸收。瘤细胞首先侵蚀骨松质,逐渐破坏骨皮质。X线片上可见骨质缺损,受累的骨组织可发生自发性骨折或脊柱塌陷。瘤组织广泛增生可引起骨质疏松。晚期,瘤细胞可浸润至软组织,并可侵犯脾、肝、肾、肺和淋巴结等。
在约半数以上的病人骨髓瘤侵犯肾,引起骨髓瘤肾病。肾体积正常或轻度肿大,色苍白,晚期因肾间质纤维化体积缩小。镜下见肾间质内有多数异常的浆细胞和慢性炎性细胞浸润。肾远曲小管和集合管内有蛋白管型,有些均匀红染,有些呈分层状或颗粒状,内含免疫球蛋白、κ或λ轻链、白蛋白等。管型周围有巨噬细胞形成的多核巨细胞环绕。肾小管上皮细胞常萎缩、坏死。由于骨组织破坏,血钙增高,可引起肾组织内转移性钙化。继发感染可引起肾盂肾炎。
【临床病理联系】
多发性骨髓瘤起病缓慢,早期多无明显症状。骨髓瘤细胞破坏骨组织可引起骨痛、骨质疏松和病理性骨折;破坏骨髓内造血组织可引起贫血、白细胞减少和血小板减少。瘤细胞产生的异常免疫球蛋白影响凝血因子和血小板功能,导致凝血障碍。加之血小板减少,病人常有出血倾向。骨髓瘤细胞产生的Ig过多及Ig聚合可使血液粘度增高,引起血粘度过高综合征,表现为紫癜、视网膜出血、出血时间延长等。血粘度过高影响血液循环引起组织缺氧,以脑、眼、肾、肢端最明显,可引起头昏、眩晕、意识障碍等神经症状。肾损伤可引起肾功能不全。由于异常的免疫球蛋白明显增多,正常的免疫球蛋白减少,病人免疫功能降低,容易并发感染,常发生肺炎和肾盂肾炎。感染和肾功能衰竭是造成死亡的主要原因。
第七节 恶性组织细胞增生症
恶性组织细胞增生症(malignant histiocytosis)是组织细胞及其前身细胞进行性恶性增生引起的一种全身性疾病。可发生于任何年龄,较多见于儿童和青壮年。男性比女性多见,约为2~3:1。临床表现为不规则发热,乏力,消瘦,全身淋巴结肿大,肝、脾肿大,有时可有皮肤损害。晚期可出现黄疸、贫血、白细胞和血小板减少和进行性衰竭。
【病理变化】
恶性组织细胞增生症主要侵犯淋巴结、脾、肝、骨髓和皮肤,全身其他器官和组织如肺、肾、心、胃、肠、胰和中枢神经系统等都可受累。各病例累及的器官和组织多少不等,即使在同一器官或组织内,病变分布情况也很不一致。一般至少有两个以上的不同器官和组织同时受累,以淋巴结、脾、肝、骨髓为最常见也最严重。淋巴结和肝、脾肿大。镜下可见淋巴结、肝、脾等组织内异常的肿瘤性组织细胞呈弥漫性增生。肿瘤细胞分化程度很不一致,有些分化程度较高,几乎接近正常的组织细胞。有些分化差,呈明显的异型性,肿瘤细胞大小、形态不一。有些肿瘤细胞体积大,核大,圆形或椭圆形,染色质稀疏,染色较淡,胞浆丰富,伊红色,有吞噬现象。胞浆内可见被吞噬的红细胞、淋巴细胞、浓缩的细胞核、细胞残屑、含铁血黄素和脂类物质。有些瘤细胞体积大,核也大,可呈双核或多核,核膜厚,核染色质分布不均匀,核仁大而明显,呈嗜碱性。还有些瘤细胞体积较小,核相对较大,染色很深,有时呈折叠状,胞浆伊红色。这些较小的肿瘤细胞多无吞噬现象。此外,常见多数典型和不典型的核分裂像。肿瘤性组织细胞吞噬红细胞的现象是恶性组织细胞增生症的重要特征。
应用组织化学和免疫组织化学反应显示肿瘤细胞内非特异性酯酶、α-萘酚醋酸酯酶、酸性磷酸酶和溶菌酶皆为阳性,说明这些肿瘤细胞来源于单核吞噬细胞系统的组织细胞。
瘤细胞在体内各组织和器官内的分布情况不同,在淋巴结内的瘤细胞多浸润于包膜下淋巴窦和髓质淋巴窦,可逐渐浸润至髓质、淋巴索和皮质。瘤细胞呈弥漫性浸润,不形成团块。除肿瘤性组织细胞增生外,淋巴结内并常有多少不等的浆细胞浸润,部分可有出血和坏死。早期淋巴结的包膜和结构多完整,晚期瘤细胞可浸润淋巴结包膜,淋巴结结构可部分或全部被破坏。脾内瘤细胞主要浸润于红髓,有时由于脾窦高度淤血,可将瘤细胞掩盖。晚期,瘤细胞大量增生可破坏脾小体。肝内瘤细胞主要浸润于汇管区和肝细胞索之间,甚似Kupffer细胞增生。瘤细胞大量增生可破坏邻近的肝细胞。骨髓内的瘤细胞多呈灶性分布。有些病人可有皮肤损害,表现为丘疹样病损,瘤细胞多浸润于真皮皮肤附件周围。
【临床病理联系】
本病可累及全身各器官,病变分布和范围各不相同,因此临床表现多样。淋巴结、肝、脾因大量瘤细胞浸润而肿大。骨髓内造血组织被瘤组织破坏,异型组织细胞常吞噬红细胞,故患者常有全血细胞减少和严重的进行性贫血,并常有出血和各种继发感染。晚期,瘤细胞损伤肝细胞可引起黄疸。肺部受累可引起咳嗽,咳血,严重者可出现呼吸衰竭。消化道的病变可引起腹泻和消化道出血。中枢神经受累时,根据病变部位不同可引起脑膜炎、失明、尿崩症、眼球突出和截瘫等。本病起病较急,进展迅速,预后较差。
第十二章 泌尿系统疾病
泌尿系统包括肾、输尿管、膀胱和尿道。其功能是将人体代谢过程中产生的废物和毒物通过尿的形式排出体外以维持机体内环境的相对稳定。肾是尿生成的重要器官。不仅可将体内的代谢废物和毒物排出体外,并且对调节体内水与电解质和维持血液的酸碱平衡都有很重要的作用。此外,肾还具有内分泌作用,可分泌重要的激素,如肾素、促红细胞生成素、前列腺素、1,25-二羟胆钙化醇(Vit·D),参与调节血压、红细胞的生成和钙的吸收。
肾的解剖生理单位称为肾单位,由肾小球和肾小管组成。每个肾约有130万个肾单位。在正常情况下,肾单位交替地进行活动,因此肾具有很大的储备代偿能力。
肾小球由毛细血管丛和肾球囊构成,是血浆滤过的器官。肾小球毛细血管壁分3层,中间为基底膜,内侧有内皮细胞覆盖,外侧为脏层上皮细胞。毛细血管基底膜厚约320nm,可分为三层,中间为致密层,内侧和外侧各为内疏松层和外疏松层。毛细血管基底膜内面由一层扁平的内皮细胞覆盖。内皮细胞胞浆很薄,布满许多直径约70~100nm的小孔。脏层上皮细胞(又称足细胞)在基底膜外侧,胞浆丰富形成许多细长的分枝状突起称为足突。上皮细胞由这些足突附着于基底膜外疏松层。足突之间形成许多间隙,宽约20~30nm,称为滤过隙。距基底膜表面约60nm,在相邻的足突之间有一层薄膜称为滤过隙膜。毛细血管壁包括内皮细胞、基底膜、上皮细胞,共同组成肾小球的滤过膜(图12-1)。肾小球的滤过除与毛细血管的结构和滤过物质的分子大小有关外,并与基底膜的生物化学组成及其电荷有关。基底膜主要由Ⅳ型胶原和一些糖蛋白如层连蛋白(laminin)、纤维连接蛋白(fibronectin)和多聚阴离子多糖蛋白(polyanionic proteoglcans)等组成。其中尤其是硫酸类肝素多糖蛋白(heparan sulfate proteoglycan)带大量负电荷分布于内、外疏松层。此外,在毛细血管内皮细胞和脏层上皮细胞表面也有带负电荷的唾液酸糖蛋白(sialoglycoprotein)。肾小球的负电荷可阻止血液中带负电荷的分子如白蛋白滤过。当肾小球多聚阴离子减少时滤过的蛋白质可增加。
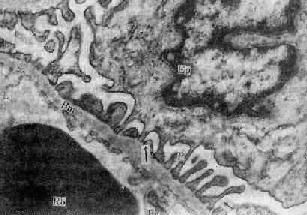
图12-1 正常肾小球滤过膜
Bm基底膜;En内皮细胞;Ep上皮细胞;L毛细血管腔;P上皮细胞足突;Rb红细胞短箭头为滤过隙;长箭头为滤过隙膜
在肾小球毛细血管之间有少量组织支持毛细血管网,并将毛细血管联系在一起,称为肾小球系膜(mesangium),由系膜细胞和系膜基质组成。系膜基质充填在各叶毛细血管之间。系膜细胞散在于系膜基质内(图12-2)。系膜细胞有收缩功能,可参与调节肾小球毛细血管的血流。系膜细胞还具有吞噬功能,可吞噬进入肾小球的大分子物质。系膜细胞可产生血管活性物质、细胞因子(如IL-1,IL-6,TNF等)和生长因子(PDGF,TGF等),并可产生系膜基质和胶原纤维,对清除肾小球滤过的物质以及与肾小球炎症和损伤时的增生和修复都有重要关系。
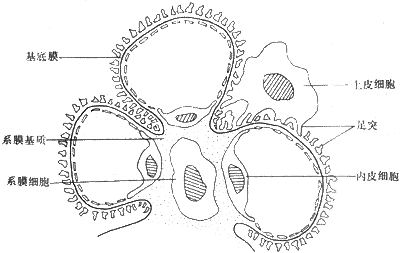
图12-2 肾小球小叶示意图
示肾小球毛细血管和系膜的结构及其相互关系
肾的各组成部分之间有密切联系。损伤时常相互影响,一部分的病变可引起其它部分的损害。因此肾疾病晚期往往各个部分都被破坏。肾小球不能再生,损伤后只能由存留的肾单位肥大来代偿损失的功能,所以肾小球发生严重的弥漫性病变时可造成严重后果。肾小管的再生能力很强,发生损伤时,如及时再生可恢复功能。肾的代偿储备能力很大,因此肾功能障碍往往在病变比较严重时才表现出来,有些已到疾病晚期,所以注意早期可能出现的症状非常重要。
泌尿系统的疾病种类很多,结合病因和病变发生的主要部位,主要可分为以下几类:①炎症:包括变态反应性炎如肾小球肾炎、泌尿道的感染如肾结核、肾盂肾炎、膀胱炎、尿道炎等;②代谢性疾病如糖尿病性肾硬化;③血管疾病如高血压性肾硬化;④中毒性疾病如汞中毒,磺胺药物等中毒引起的急性肾小管坏死等;⑤尿路阻塞如泌尿道结石和肾盂积水等;⑥先天性畸形如多囊肾、马蹄肾、输尿管瓣膜等;⑦遗传性疾病如遗传性肾炎;⑧肿瘤如肾细胞癌,膀胱乳头状瘤和膀胱移形细胞癌等。
第一节 肾小球肾炎
肾小球肾炎(glomerulonephritis)是以肾小球损害为主的变态反应性炎症,是一种比较常见的疾病。临床表现主要有蛋白尿,血尿,水肿和高血压等。早期症状常不明显,容易被忽略,发展到晚期可引起肾功能衰竭,严重威胁病人的健康和生命,是引起肾功能衰竭最常见的原因。肾小球肾炎可为原发性和继发性。原发性肾小球肾炎指原发于肾的独立性疾病。病变主要累及肾。继发性肾小球肾炎的肾病变是其他疾病引起的,或肾病变是全身性疾病的一部分,如红斑狼疮性肾炎、过敏性紫癜性肾炎等。此外,血管病变如高血压、代谢疾病如糖尿病等都可引起肾小球病变。一般所称肾小球肾炎若不加说明常指原发性肾小球肾炎。部分继发性肾小球疾病在其他有关章节叙述。本节仅介绍原发性肾小球肾炎。
【病因和发病机制】
肾小球肾炎(简称肾炎)的病因和发病机制虽然尚水完全明了,但近年来的研究对阐明肾炎的病因和发病机制取得了很大进展。大量实验和临床研究证明肾炎的大多数类型都是抗原抗体反应引起的免疫性疾病。细胞免疫可能对某些肾炎的发病也有一定作用。
(一)引起肾小球肾炎的抗原
引起肾小球肾炎的抗原物质有些还不了解,已知的大致可分为内源性和外源性两大类。
1.内源性抗原
(1)肾小球本身的成分:肾小球基底膜的成分如层连蛋白和Goodpasture抗原(Ⅳ型胶原羧基端球状部的一种多肽),肾小球毛细血管上皮细胞的Heymann抗原(一种膜糖蛋白),内皮细胞膜抗原,系膜细胞膜抗原等。
(2)非肾小球抗原:核抗原,DNA,免疫球蛋白,免疫复合物,肿瘤抗原,甲状腺球蛋白抗原等。
2.外源性抗原
(1)感染的产物:细菌如链球菌,葡萄球菌,肺炎球菌,脑膜炎球菌,伤寒桿菌等。病毒如乙型肝炎病毒,麻疹病毒,EB病毒等。霉菌如白色念珠菌等和寄生虫如疟疾,Manson血吸虫,丝虫等。
(2)药物如青霉胺,金和汞制剂等。
(3)异种血清,类毒素等。
各种不同的抗原物质引起的抗体反应和形成免疫复合物的方式和部位不同,与肾小球肾炎的发病和引起的病变类型有密切关系。
(二)肾小球肾炎的免疫发病机制
免疫复合物形成引起肾小球肾炎基本上有两种方式:①抗体与肾小球内固有的不溶性肾小球抗原或植入在肾小球内的非肾小球抗原,在肾小球原位结合形成免疫复合物;②血液循环内形成的可溶性抗原抗体复合物沉积于肾小球。
1.肾小球原位免疫复合物形成抗体与肾小球内固有的抗原成分或植入在肾小球内的抗原结合,在肾小球原位直接反应,形成免疫复合物,引起肾小球损伤。近年来的研究证明,肾小球原位免疫复合物形成在肾小球肾炎的发病中起主要作用。由于抗原性质不同所引起的抗体反应不同,可引起不同类型的肾炎。
(1)肾小球基底膜抗原:肾小球基底膜本身的成分为抗原,机体内产生抗自身肾小球基底膜抗体,这种自身抗体直接与肾小球基底膜结合形成免疫复合物(图12-3)。用免疫荧光法可见免疫复合物沿肾小球毛细血管基底膜沉积呈连续的线形荧光(图12-4)。肾小球基底膜抗原的性质可能是基底膜内Ⅳ型胶原羧基端非胶原区的一种多肽。关于机体产生抗自身肾小球基底膜抗体的原因目前还不完全明了。可能在感染或某些因素的作用下,基底膜的结构发生改变而具有抗原性,可刺激机体产生自身抗体。或某些细菌、病毒或其他物质与肾小球基底膜有共同抗原性,这些抗原刺激机体产生的抗体可与肾小球毛细血管基底膜起交叉反应。抗肾小球基底膜抗体引起的肾炎称为抗肾小球基底膜性肾炎,是一种自身免疫性疾病。这类肾炎在人类较少见,约占人类肾小球肾炎的5%。
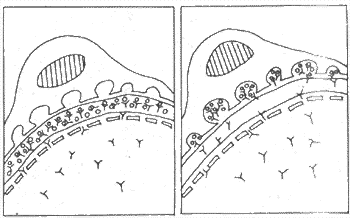
图12-3 肾小球肾炎原位免疫复合物形成机制示意图
左图:抗肾小球基底膜性肾小球肾炎 右图:非肾小球基底膜性抗原包括其他肾小球抗原或植入性抗原在肾小球内与抗体结合
○抗原,Y抗体,![]() 抗原抗体复合物
抗原抗体复合物
图12-4 抗肾小球基底膜性肾炎
免疫复合物沿肾小球毛细血管基底膜沉积,呈连续的线形荧光免疫荧光染色FITC-IgG
(2)其他肾小球抗原:除肾小球基底膜外,肾小球内其他抗原成分如系膜细胞膜抗原Thy-1和上皮细胞的Heymann抗原等也可引起肾小球原位免疫复合物形成。典型的代表为实验性大鼠的Heymann肾炎。用肾小管刷状缘抗原免疫大鼠后,大鼠体内产生抗肾小管刷状缘抗体,并引起肾小球肾炎。目前已知这种刷状缘抗原即Heymann抗原,是一种分子量为330kD的糖蛋白(gp330),主要位于近曲小管刷状缘和肾小球。肾小球的gp330由脏层上皮细胞合成,合成后集中在上皮细胞足突底部表面与毛细血管基底膜相邻处。抗体与足突底部的gp330抗原结合,在毛细血管表面形成多数小丘状免疫复合物,免疫荧光染色呈不连续的颗粒状荧光(图12-5)。电子显微镜下可见肾小球毛细血管基底膜表面上皮细胞下有多数小堆状电子致密物沉积。
图12-5 膜性肾小球肾炎
免疫复合物沉积在肾小球毛细血管表面,呈不连续的颗粒状荧光免疫荧光染色FITC-IgG
(3)植入性抗原:非肾小球抗原可与肾小球内的成分结合,形成植入性抗原而引起抗体形成。抗体与植入抗原在肾小球内原位结合形成免疫复合物引起肾小球肾炎。
可形成肾小球植入抗原的非肾小球抗原可为内源性或外源性。例如带正电荷的非肾小球抗原或抗体,可与肾小球内带负电荷的成分如基底膜表面的硫酸类肝素、多糖蛋白或系膜细胞表面的阴离子结合形成植入抗原。免疫球蛋白,聚合的IgG等大分子物质常在系膜内沉积与系膜结合形成植入抗原。此外,细菌的产物如甲组链球菌产生的endostreptosin、病毒、寄生虫等感染产物和某些药物都可能与肾小球内的成分结合形成植入抗原。大多数植入抗原引起的肾小球肾炎,用免疫荧光法检查可见免疫复合物在肾小球内呈不连续的颗粒状荧光(图12-3右)。
2.循环免疫复合物沉积引起循环免疫复合物的抗原为非肾小球性,即不属于肾小球的组成成分。这些抗原可以是外源性的,如感染产物、异种蛋白、药物等。也可以是内源性的,如DNA、甲状腺球蛋白及肿瘤抗原等。这些抗原在机体内产生的相应抗体,对肾小球的成分无免疫特异性。抗原抗体在血液循环内结合,形成抗原抗体复合物。这些抗原抗体复合物随血液流经肾时,在肾小球内沉积引起肾小球损伤。应用电子显微镜可见肾小球内有电子致密物质沉积。用免疫荧光法检查可见免疫复合物在肾小球内呈颗粒状荧光(图12-6)。
图12-6 肾小球肾炎循环免疫复合物沉积机制示意图
○抗原;Y抗体;![]() 抗原抗体复合物
抗原抗体复合物
抗原、抗体和免疫复合物在肾小球内沉积的部位与免疫复合物的大小,抗原、抗体和免疫复合物的电荷有关。含大量阳离子的抗原容易通过肾小球基底膜,在基底膜外侧上皮细胞下形成免疫复合物。含大量阴离子的大分子物质不易通过基底膜,往往在内皮细胞下沉积,或被吞噬清除,不引起肾炎。接近中性的分子形成的免疫复合物往往容易沉积于系膜内。此外,抗原和抗体的性质,肾小球的结构和形态,如电荷、通透性、血液动力学以及系膜细胞和单核巨噬细胞的功能等都与免疫复合物形成的部位和所引起的肾组织病变类型有密切关系。
以上引起肾炎的两种途径,即肾小球原位免疫复合物形成和循环免疫复合物形成并非完全互不相关。两者既可单独进行,也可共同作用引起肾小球肾炎。两种发病机制引起的肾炎都可表现为急性或慢性过程,病变也都可有轻有重。这一方面与抗原和抗体的性质、数量、免疫复合物沉积的量和持续的时间有关。另方面也取决于机体的免疫状态和反应性等内在因素。
关于细胞免疫在肾小球肾炎发生中的作用,由于有些病人或实验性肾炎组织中无免疫复合物,但可见巨噬细胞或淋巴细胞反应,故有人认为,这些肾炎的发生可能与T细胞介导的细胞免疫有关。近年来有实验证明,T细胞可不依赖抗体引起肾炎。
(三)引起肾小球肾炎的介质
免疫复合物在肾小球内沉积后,可通过不同的机制引起肾小球损伤。引起肾小球损伤的主要介质包括抗体、补体、中性粒细胞、单核巨噬细胞、血小板、系膜细胞和凝血系统等。
1.抗体通过各种方式沉积在肾小球内的抗体可以在没有补体或炎症细胞的参与下,单独引起肾小球损伤。实验证明豚鼠的一种抗肾小球基底膜抗体和有些不固定补体的抗肾小球上皮细胞膜的单克隆抗体,可作用于上皮细胞膜引起上皮细胞的改变,使上皮细胞与基底膜脱离或损伤基底膜引起肾炎。
2.补体免疫复合物结合并激活补体。活化补体可通过两种方式:①吸引中性粒细胞和②补体的末端成分(C5b~9)引起肾小球损伤。
补体成分C3a,C5a具有过敏毒素作用使血管通透性增加。C3a,C5a和C567具有化学趋向性可吸引中性粒细胞。C3b可增强粒细胞的吞噬作用和促进免疫吸附。中性粒细胞粘附于血管壁,释放炎症介质尤其是蛋白酶和氧代谢活性产物破坏毛细血管内皮细胞和基底膜。
补体的末端成分(C5b~9)形成膜攻击复合物可在没有中性粒细胞参与下引起肾小球损伤。C5b~9可刺激肾小球脏层上皮细胞产生氧代谢活性产物和蛋白酶等炎症介质损伤基底膜,并可刺激系膜细胞产生蛋白酶、氧自由基、白细胞介素-1和前列腺素等引起肾小球损伤。
3.中性粒细胞中性粒细胞除通过补体成分的化学趋向性作用在肾小球聚集引起炎症外,有些细胞损伤后释放的血小板活化因子、血小板源性生长因子、白细胞三烯B4等也具有吸引中性白细胞的作用。
4.单核细胞和巨噬细胞有些肾炎时肾小球内有多数单核细胞浸润。活化的单核细胞可产生多种生物活性物质如蛋白酶、白细胞三烯、前列腺素、IL-1、血小板源性生长因子和凝血因子等与肾小球炎症的发生和发展有关。
5.血小板有些肾炎时通过补体的作用,血小板在肾小球内集聚释放血栓素A2,血小板活化因子,花生四烯酸代谢产物和生长因子等可促进白细胞释放蛋白分解酶和促使系膜细胞增生引起肾小球炎症。
6.系膜细胞 在免疫复合物、补体、C5b~9、内毒素和生长因子等作用下,系膜细胞增生并被活化。活化的系膜细胞可产生许多炎症介质如蛋白酶,氧代谢活性产物,IL-1及生长因子等可不依赖中性粒细胞引起肾小球炎症。
7.凝血系统内皮细胞损伤,基底膜胶原暴露激活Ⅻ因子。活化的Ⅻ因子可激活凝血系统、激肽系统及纤维蛋白溶酶系统引起炎症。此外,内皮细胞和单核巨噬细胞产生的促凝血因子可促使渗入肾球囊的纤维蛋白原凝集,刺激细胞增生,与肾小球病变的发展,尤其是新月体的形成有重要关系。
【肾小球肾炎的分类】
肾小球肾炎的命名和分类方法很多,分类的基础和依据各不相同,意见也不完全一致。近三十余年来通过开展肾穿刺活检,对肾炎的病理变化和病变发展过程的认识有了很大提高。肾炎的病理变化与临床发展有密切联系,肾小球肾炎的病理分类对临床治疗和预后有很大帮助,已在各国普遍应用。本节主要根据1982年WHO美国J.Churg教授主持制定的肾小球疾病的病理分类,介绍原发性肾小球肾炎的主要类型(表12-1)及其特点和病理变化。
表12-1 原发性肾小球肾炎的分类
| 1.肾小球轻微病变 |
| 2.局灶性/节段性肾小球硬化 |
| 3.局灶性/节段性肾小球肾炎 |
| 4.弥漫性肾小球肾炎 |
| (1)膜性肾小球肾炎(膜性肾病) |
| (2)系膜增生性肾小球肾炎 |
| (3)毛细血管内增生性肾小球肾炎 |
| (4)膜性增生性肾小球肾炎(系膜毛细血管性肾小球肾炎)Ⅰ型及Ⅲ型 |
| (5)致密沉积物性肾小球肾炎(致密沉积物病;膜性增生性肾小球肾炎Ⅱ型) |
| (6)新月体性(毛细血管外增生性)肾小球肾炎 |
| 5.未分类肾小球肾炎 |
根据肾炎时肾组织病变的范围可将肾小球肾炎分为局灶性和弥漫性。病变累及肾组织中全部或绝大部分肾小球者称为弥漫性肾小球肾炎;病变仅累及少数或部分肾小球者称为局灶性肾小球肾炎。根据肾小球的病变情况,每个肾小球的病变可分为球性和节段性。整个肾小球皆受累者称为球性,病变仅累及肾小球的一部分或少数毛细血管袢者称为节段性。
肾小球肾炎的病变类型较多,临床表现也不相同。肾炎的病变类型与临床表现虽有密切关系,但不完全平行。相似的症状可由不同的病变引起,相似的病变也可引起不同的症状。此外,肾炎的临床表现尚与病变的严重程度、发病机制、持续时间、发病急缓等都有关系。一般可将肾炎的临床表现大致分为以下几个类型。
(1)急性肾炎综合征:起病急,常突然出现血尿、程度不同的蛋白尿、少尿、水肿、高血压,肾小球滤过率降低。
(2)快速进行性肾炎综合征:突然或逐渐出现血尿、少尿、蛋白尿、贫血,快速进展为肾功能衰竭。
(3)复发性或持续性血尿:起病缓慢或急骤,常表现为肉眼或镜下血尿,可伴有轻度蛋白尿。一般没有其他肾炎症状。
(4)慢性肾炎综合征:起病缓慢,逐渐发展为慢性肾功能不全,可伴有蛋白尿、血尿和高血压。
(5)肾病综合征:表现为大量蛋白尿、严重水肿、低蛋白血症,并常有高脂血症。
肾小球肾炎的分类比较复杂,为了便于理解,现根据各型肾炎的病变特点简介如下。
一、弥漫性毛细血管内增生性肾小球肾炎
弥漫性毛细血管内增生性肾小球肾炎(diffuse endocapillary proliferative glomerulonephritis)病变为弥漫性,两侧肾几乎全部肾小球皆受累。这种肾炎常发生于感染以后,故有感染后肾小球肾炎之称。最常见的为A组乙型溶血性链球菌感染,其中以12型、4型、1型及49型与肾炎的关系最为密切。一般发生在链球菌感染后1~3周,是链球菌感染引起的变态反应性疾病,称为链球菌感染后肾炎。除链球菌外,其他细菌如葡萄球菌、肺炎球菌和某些病毒及寄生虫等也可引起这种类型的肾炎。这种肾炎多见于儿童,成人也可发生,但病变往往比儿童严重。一般发病较急,临床主要表现为急性肾炎综合征。
【病理变化】
病变为弥漫性,两侧肾同时受累。病变进展较快,主要变化为肾小球内细胞增生。早期,肾小球毛细血管充血,内皮细胞和系膜细胞肿胀增生并有少量中性粒细胞浸润。毛细血管通透性增加,血浆蛋白质可以滤过而进入肾球囊。因此,病人的尿液中常有蛋白、红细胞及白细胞。轻型病人,病变可不再发展,以后逐渐痊愈;比较严重的病例,病变继续发展,肾小球内细胞增生加重。增生的细胞主要为系膜细胞和内皮细胞。增生细胞压迫毛细血管,使毛细血管腔狭窄甚至闭塞,肾小球呈缺血状(图12-7)。此外,肾小球内有多数炎性细胞浸润,主要为中性粒细胞,有时并有少数嗜酸性粒细胞、单核细胞、红细胞、浆液和纤维素性渗出液。镜下,肾小球内细胞数量增多,肾小球体积增大。病变严重时,毛细血管腔内可有血栓形成,毛细血管壁可发生纤维素样坏死。坏死的毛细血管袢破裂出血,大量红细胞进入肾球囊及肾小管腔内,可以引起明显的血尿。不同的病例病变表现形式可能不同。有的以渗出为主,称为急性渗出性肾小球肾炎。有些病变严重,肾小球毛细血管袢坏死,有大量出血者称为出血性肾小球肾炎。
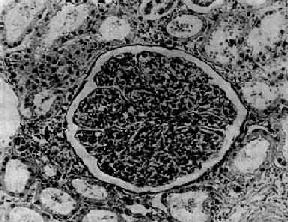
图12-7 弥漫性毛细血管内增生性肾小球肾炎
肾小球内细胞数量增多,系膜细胞和内皮细胞增生并有少量中性白细胞浸润,毛血管腔狭窄
上皮细胞一般无明显增生,少数严重的病例肾小球的壁层和脏层上皮细胞可增生,形成新月体。这种病变容易引起肾小球纤维化。如数量少,对功能影响不大。如病变广泛,可发展为新月体性肾炎。
在电子显微镜下可见肾小球系膜细胞和内皮细胞增生肿胀。基底膜和脏层上皮细胞间有致密物质沉积。这些沉积物大小不等,有的很大,在基底膜表面呈驼峰状或小丘状(图12-8,图12-9)。沉积物表面的上皮细胞足突多消失。基底膜变化不明显有时边缘稍不规则。沉积物一般在发病后几天就可出现,在4~6周内消失。有时基底膜内侧内皮细胞下和系膜内也可见小型沉积物。

图12-8 毛细血管内增生性肾小球肾炎
电镜下见肾小球毛细血管基底膜表面上皮细胞下有多数驼峰状电子致密沉积物
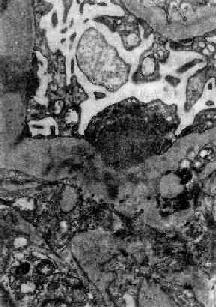
图12-9 毛细血管内增生性肾小球肾炎
电镜下见驼峰状沉积物位于毛细血管基底膜表面。沉积物表面有上皮细胞覆盖,上皮细胞足突消失
免疫荧光法检查显示,在肾小球毛细血管壁表面有免疫球蛋白和补体沉积(主要为IgG和C3),呈颗粒状荧光。系膜内也可有类似沉积物。
肾小球的病变可引起相应的肾小管缺血,肾小管上皮细胞常有浊肿、玻璃样变和脂肪变等。管腔内含有从肾小球滤过的蛋白、红细胞、白细胞和脱落上皮细胞。这些物质在肾小管内凝集,形成各种管型,如蛋白管型、透明管型、细胞管型(如红细胞、白细胞或上皮细胞管型)、颗粒管型。
肾间质内常有不同程度的充血、水肿和少量淋巴细胞、中性粒细胞浸润。
肉眼观,早期变化不明显。以后肾轻度或中度肿大、充血、包膜紧张、表面光滑、色较红,故称大红肾。若肾小球毛细血管破裂出血,肾表面及切面可见散在的小出血点如蚤咬状,称蚤咬肾。切面可见皮质由于炎性水肿而增宽,条纹模糊与髓质分界明显。
【临床病理联系】
这种肾炎的主要临床症状为尿的变化,水肿和高血压。
1.尿的变化由于肾小球毛细血管损伤,通透性增加,故常有血尿、蛋白尿、管型尿等。
(1)血尿:血尿常可反映肾小球毛细血管损伤的情况。轻度血尿需用显微镜才能发现。严重的血尿,肉眼可见尿呈鲜红色。有时尿中红细胞溶解,血红蛋白在酸性尿中转变成酸性血红素,使尿呈棕红色。
(2)蛋白尿:蛋白尿的程度不同,一般不很严重,但少数病人尿中可有大量蛋白质。
(3)管型尿:在肾小管内凝集形成的管型随尿液排出,尿液内可出现各种管型,称为管型尿。
(4)少尿:由于肾小球细胞增生肿胀,压迫毛细血管,致管腔狭小,肾血流受阻,肾小球滤过率降低,而肾小管再吸收无明显障碍,可引起少尿,致水钠在体内潴留。严重者并可有含氮代谢产物潴留,引起氮质血症。
2.水肿病人常有轻度或中度水肿,往往首先出现在级织疏松的部位如眼睑。水肿的原因主要是由于肾小球滤过减少,而肾小管再吸收功能相对正常,引起水钠潴留。此外,也可能与变态反应所引起的全身毛细血管痉挛和通透性增加有关。
3.高血压病人常有轻至中度高血压。过去认为这种肾炎时的高血压与肾小球毛细血管阻塞,肾组织缺血引起肾素分泌增加有关。但病人血中肾素浓度多在正常范围之内。因此高血压的主要原因可能与水钠潴留引起的血量增加有关。严重的高血压可导致心力衰竭及高血压性脑病。
【结局】
这种肾炎的预后与年龄和病因有一定关系。儿童链球菌感染后肾小球肾炎的预后很好,95%以上可在数周或数月内症状消失,病变消退,完全恢复。少数病人病变消退较慢,肾小球系膜增生,可持续数月甚至1~2年。临床上,病人常有迁延性蛋白尿和复发性血尿。有时无明显症状,成为隐匿性肾炎。这种病变大多以后仍可消退,恢复正常。少数病人(约占1%~2%)临床症状消失,但病变持续不退,以后症状可反复,逐渐发展为慢性硬化性肾小球肾炎。极少数病人病变严重,发展较快,同时有明显的肾球囊上皮细胞增生,形成大量新月体,可发展为新月体性肾小球肾炎。这些病人常迅速发生急性肾功能衰竭,预后差。还有极少数(<1%)病人病变严重,发展迅速,在短期内发生肾功能衰竭、心力衰竭或高血压性脑病。
一般成人患感染后肾小球肾炎者预后较差,发生肾功能衰竭和转变为慢性肾炎者较多。此外,由其他感染引起的肾炎转变为慢性肾小球肾炎者,比链球菌感染后肾炎转为慢性者多见,预后也较差。
二、弥漫性系膜增生性肾小球肾炎
弥漫性系膜增生性肾不球肾炎(diffuse mesangial proliferative glomerulonephritis)的病变特点为弥漫性肾小球系膜增生。多见于青少年,我国及东方国家比西方国家多见。本病可为原发性,也可在一些全身性疾病时发生,如系统性红斑狼疮、过敏性紫癜等。有些迁延性毛细血管内增生性肾小球肾炎病变持续不退,可表现为系膜增生性肾小球肾炎。临床可表现为肉眼或镜下血尿或蛋白尿,或两者兼有。少数表现为肾病综合征。
【病理变化】
主要病变为肾小球系膜细胞和基质增生,系膜区增宽(图12-10,图12-11)。毛细血管壁无明显变化,管腔通畅。系膜内可有少数单核细胞和中性粒细胞浸润。病变严重者可引起系膜硬化。
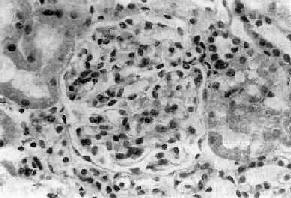
图12-10 弥漫性系膜增生性肾小球肾炎
肾小球系膜区略增宽,系膜细胞数量增多
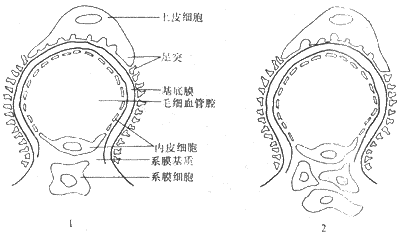
图12-11正常肾小球毛细血管及系膜增生性肾小球肾炎示意图
1.正常肾小球毛细血管及系膜示意图 2.系膜增生性肾小球肾炎示系膜增生
电子显微镜检查可见除肾小球系膜增生外,系膜内并有电子致密物沉积。有时肾小球基底膜表面上皮细胞下、内皮细胞下也可见小形致密沉积物。
免疫荧光法检查显示,系膜区内有免疫复合物沉积,主要为IgM,有时可同时伴有C3沉积。有些病例系膜区内免疫复合物主要为IgG、IgA及C3。
【临床病理联系】
病变主要累及系膜,早期症状多不明显,仅有轻度蛋白尿或复发性血尿,容易被忽略。系膜内沉积的免疫复合物与临床症状有一定联系。有IgM沉积的病人多表现为肾病综合征。同时有IgG、IgA及C3沉积者多伴有血尿。
【结局】
一般病变可及时消退,预后较好。有时病变可持续2~3年,以后仍可消退。部分病人病变严重,不能及时消退,继续发展可导致系膜硬化和肾小球硬化。晚期可发展为硬化性肾小球肾炎和慢性肾功能不全。
弥漫性新月体性肾小球肾炎(diffuse crecsentic glomerulonephritis)又称毛细血管外增生性肾小球肾炎(extracapillary glomerulonephritis),比较少见,大多见于青年人和中年人,儿童与老年人也可发生,但较少见。病变特点为肾小球内有大量新月体形成。病变严重,进展很快。主要症状为血尿,并迅速出现少尿、无尿、高血压和氮质血症。如不采取措施,常在数周至数月内发生肾功能衰竭,死于尿毒症,故又称快速进行性肾小球肾炎(rapidly progressive glomerulonephritis)。这种肾炎可与其他肾小球疾病伴发,如严重的毛细血管内增生性肾小球肾炎,肺出血肾炎综合征(Goodpasture综合征)或系统性红斑狼疮,过敏性紫癜等。但多数原因不明,为原发性。
【病理变化】
病变为弥漫性。镜下可见大部分肾小球内有新月体形成。新月体主要由肾小球上皮细胞增生和渗出的单核细胞组成。肾小球上皮细胞主要是壁层上皮细胞增生显著,堆积成层,在肾球囊内毛细血管丛周围呈新月形或环状,故称为新月体或环状体(图12-12)。新月体形成很快,可在发病后数日内形成。增生的上皮细胞间可见红细胞、中性粒细胞和纤维素性渗出物。有时可见球丛毛细血管发生纤维素样坏死和出血。肾球囊内的纤维蛋白可刺激上皮细胞增生,是促使新月体形成的原因。系膜和内皮细胞也增生,但一般程度较轻。早期新月体主要由增生的上皮细胞和单核细胞组成,称为细胞性新月体。新月体内增生的上皮细胞之间逐渐出现新生的纤维细胞,以后纤维组织逐渐增多形成纤维-细胞性新月体。最后新月体内的细胞和渗出物完全由纤维组织替代,成为纤维性新月体。
图12-12 新月体性肾小球肾炎
肾球囊壁层上皮细胞增生,其间有单核巨噬细胞浸润形成新月体,肾球囊腔被阻塞
新月体形成后,一方面压迫毛细血管丛,另方面肾球囊增厚与球丛粘连,使肾球囊腔闭塞,肾小球的结构和功能严重破坏,影响血浆从肾小球滤过,最后毛细血管丛萎缩、纤维化,整个肾小球纤维化玻璃样变,功能丧失。
电镜下,可见肾小球毛细血管基底膜不规则增厚,部分变薄,有时在基底膜上、膜内或膜下可见电子密度高的沉积物。在增生上皮细胞形成的新月体内,可见纤维蛋白条索。有时可见肾小球毛细血管基底膜有裂孔或缺损。血液内的红细胞和纤维蛋白原可通过这些缺损进入肾球囊,促使肾小球上皮细胞增生。
免疫荧光检查结果不一,与致病原因有关。有些病例在肾小球毛细血管基底膜下呈现连续的线形荧光,可能与抗肾小球基底膜性肾炎有关。有些在肾小球基底膜上呈不规则的粗颗粒状荧光,约半数病例很少或不见荧光阳性沉积物。
除肾小球病变外,肾小管上皮细胞常有浊肿和脂肪变,腔内有蛋白凝固而形成的透明管型。由于肾小球纤维化,其所属的肾小管也萎缩消失。间质内纤维组织增生,有多数淋巴细胞,单核细胞等炎性细胞浸润。肉眼观可见肾体积增大,颜色苍白,皮质内有时可见散在的点状出血。
【临床病理联系】
快速进行性肾小球肾炎病人临床上多出现血尿、蛋白尿,迅速出现少尿,甚至无尿和氮质血症。由于这种肾炎常发生在坏死性肾小球肾炎的基础上,病变进展快,肾小球毛细血管坏死,基底膜缺损和出血,因此血尿常比较明显,蛋白尿相对较轻,水肿不明显。大量新月体形成后,阻塞肾球囊腔,血浆不能滤过,故出现少尿甚至无尿。代谢废物不能排出,在体内潴留引起氮质血症。大量肾单位纤维化,玻璃样变,肾组织缺血,通过肾素-血管紧张素的作用,可发生高血压。由于病变广泛,大量代谢产物在体内堆积,水电解质和酸碱平衡调节紊乱,最后可导致肾功能衰竭。
【结局】
快速进行性肾小球肾炎,由于病变广泛,发展迅速,预后较差,如不及时采取措施病人往往于数周至数月内死于尿毒症。预后一般与病变的广泛程度和新月体的数量有关。肾内80%~90%以上肾小球皆有新月体形成者往往不能恢复;50%~80%有新月体形成病变程度较轻者进展较慢,存留的肾小球可保留部分功能,病人可维持较长时间。近年来应用中西医结合治疗、人工肾和肾移植等对快速进行性肾不球肾炎的治疗取得了一定的效果。
附 肺出血肾炎综合征
肺出血肾炎综合征(Goodpasture syndrome)的主要病变为肺出血合并肾小球肾炎。这种肾炎大都是抗肾小球基底膜抗体引起的新月体性肾小球肾炎,是抗肾小球基底膜抗体引起的肾小球肾炎的典型代表。抗肾小球基底膜抗体可与肺泡基底膜发生交叉反应,引起肺出血。一般发病急,常为暴发性。临床上主要表现为咯血和快速进行性肾功能衰竭。本病较少见,多发生于青壮年,小儿及老年人较少,男性多于女性。
【病理变化】
肾的变化主要表现为新月体性肾小球肾炎。病变轻重不等,轻者早期肾小球毛细血管呈局灶性节段性增生及毛细血管袢纤维素样坏死。病变往往迅速发展引起大量新月体形成,其间可见单核细胞和中性粒细胞浸润。电镜下可见毛细血管基底膜局部破裂,周围有红细胞和纤维蛋白聚集。免疫荧光法检查可见免疫球蛋白和补体沿肾小球毛细血管基底膜沉积,呈连续性线形荧光。
在肺出血肾炎综合征的病人中约95%以上可在血液循环内检出抗肾小球基底膜抗体。这些抗体与肺泡基底膜及肾小管和肾球囊基底膜雹有交叉反应。虽然目前认为肺出血肾炎综合征是抗肾小球基底膜抗体引起的自身免疫性疾病,但是促使机体产生抗肾小球基底膜抗体的原因还不清楚,可能与遗传素质有关。
【临床病理联系和结局】
病人开始时往往先有肺部症状,主要表现为反复咯血,随后出现肾炎症状,主要表现为血尿、蛋白尿、少尿,常迅速发展至无尿、氮质血症和尿毒症。病人的预后与病变范围有关。如肾小球新月体形成少于80%,则预后较好,如不及时采取措施病人往往死于肺部大出血或肾功能衰竭。近年来应用血浆交换和肾移植治疗效果较好。
四、弥漫性膜性增生性肾小球肾炎
弥漫性膜性增生性肾小球肾炎(diffuse membranoproliferative glomerulonephritis)又称系膜毛细血管性肾小球肾炎(mesangiocapillary glomerulonephritis),其病变特点为弥漫性肾小球毛细血管壁增厚和系膜增生。各种年龄都可发病,但多见于青年和中年人。起病缓慢,是一种慢性进行性疾病。早期症状不明显,临床症状表现不一,常有血尿、蛋白尿。约半数起病时即表现为肾病综合征,并常有高血压和肾功能不全。有些病人的血清补体降低,故又称为低补体血症性肾小球肾炎(hypocomplementemic glomerulonephritis)。
【病理变化】
肾小球系膜细胞和基质增生。系膜区增宽,其间常有数量不等的中性粒细胞浸润。增生的系膜组织逐渐向周围毛细血管伸展,侵入毛细血管基底膜与内皮细胞之间,使毛细血管壁增厚,管腔狭窄(图12-13)。应用银染法可见增厚的毛细血管壁呈车轨状或分层状。由于系膜增生,系膜区增宽,使毛细血管丛呈分叶状。严重时可使轴心处呈结节状。病变继续发展,增生的系膜组织可环绕全部毛细血管壁,使管壁显著增厚,管腔狭小甚至阻塞。有时部分肾小球内可有新月体形成。晚期,系膜及肾小球纤维化、硬化,整个肾小球形成无结构的玻璃样小团。相应的肾小管萎缩,间质纤维组织增生,有多数淋巴细胞及单核细胞浸润。
图12-13 膜性增生性肾小球肾炎
肾小球系膜细胞增多,增生的系膜组织侵犯毛细血管,毛细血管壁增厚,腔狭小,肾小球丛呈分叶状
电镜观察可见肾小球系膜增生,毛细血管基底膜不规则增厚。增生的系膜组织沿毛细血管基底膜和内皮细胞之间向毛细血管周围部分伸展,甚至环绕全部毛细血管壁,使毛细血管壁增厚。肾小球内有大量电子致密物沉积。根据沉积物的部位,可将膜性增生性肾小球肾炎分为3型:
Ⅰ型电子致密物沉积在肾小球基底膜内侧,内皮细胞下,大小不等,常聚积成大团块状(图12-14,图12-15)。系膜内和上皮细胞下偶然也可见有少量小而不规则的沉积物。
图12-14 膜性增生性肾小球肾炎Ⅰ型示意图
肾小球系膜增生并侵入周围毛细血管,血管壁增厚,基底膜内侧内皮细胞下有电子致密沉积物
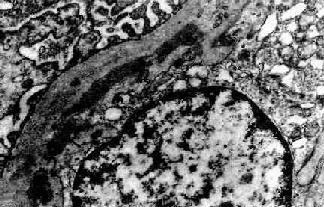
图12-15 膜性增生性肾小球肾炎 Ⅰ型
肾小球毛细血管基底膜内侧,内皮细胞下电子致密沉积物,部分上皮细胞足突消失
Ⅱ型肾小球毛细血管基底膜不规则增厚。在基底膜致密层内有高电子密度的粗大呈带状的沉积物(图12-16)。故这型肾炎又称为致密沉积物性肾小球肾炎或致密沉积物病。
图12-16 膜性增生性肾小球肾炎Ⅱ型示意图
肾小球系膜增生并侵入周围毛细血管,血管壁增厚,毛细血管基底膜致密层内有高电子密度致密沉积物,粗大呈带状
Ⅲ型肾小球毛细血管基底膜内皮细胞下和上皮细胞下都有电子致密物沉积,并可伴有基底膜断裂。
免疫荧光检查,Ⅰ型显示IgG和C3沿肾小球毛细血管和系膜区内呈颗粒状荧光,有时也有少量C1q,C4及C2。Ⅱ型时主要有大量C3沉积,一般无其他早期的补体成分和免疫球蛋白。可能说明在这种肾炎的发病中,补体主要通过替代途径激活。
【临床病理联系】
肾小球内大量C3沉积,说明肾炎发病过程中大量补体被消耗,故病人血清补体降低。
早期病变主要在系膜,血管壁变化较轻,症状不明显,或仅有轻度蛋白尿或血尿。病变逐渐发展侵犯毛细血管壁可引起肾病综合征。晚期,系膜硬化,肾小球毛细血管腔狭小或阻塞,肾小球纤维化,肾小球血流减少,可导致高血压和肾功能不全。
【结局】
病变为慢性进行性,病程长,对激素或免疫抑制剂治疗效果短暂或不明显,可逐渐发展为弥漫性硬化性肾小球肾炎。约50%的病人在10年内发生慢性肾功能衰竭。肾移植后复发率也较高,尤其是Ⅱ型膜性增生性肾炎预后更差。
五、弥漫性膜性肾小球肾炎
弥漫性膜性肾小球肾炎(diffuse membranous glomerulonephritis)的主要病变为弥漫性肾小球毛细血管基底膜增厚,常伴有大量蛋白尿,是引起肾病综合征的主要原因之一。由于肾小球无明显炎症现象,故又称为膜性肾病。起病缓慢,病程长,多见于青年和中年人,儿童患者较少。
【病理变化】
病变为弥漫性,镜下可见肾小球毛细血管壁增厚,通透性明显增加,早期病变轻,不易察觉,易与轻微病变性肾小球肾炎混淆。随病变进展,管壁增厚逐渐加重。
电镜下可见毛细血管基底膜表面,上皮细胞下,有多数细小的小丘状沉积物。基底膜表面形成许多钉状突起插入小丘状沉积物之间。银染色基底膜及钉状突起呈黑色。钉状突起与基底膜垂直相连形如梳齿(图12-17,图12-18)。免疫荧光法证实沉积物内含免疫球蛋白和补体,多为IgG和C3,沿基底膜表面呈颗粒状荧光。早期沉积物和基底膜钉状突起少而细小,以后逐渐增多、增大,基底膜增厚,钉状突起伸向沉积物表面将沉积物包围,最后大量沉积物被埋藏在增厚的基底膜内,基底膜高度增厚(图12-19)。沉积物在增厚的基底膜内逐渐溶解,使基底膜呈虫蚀状。以后这些空隙由基底膜物质充填。由于基底膜高度增厚,故毛细血管腔狭小,甚至阻塞。系膜硬化,最后整个肾小球被玻璃样变。
图12-17 膜性肾小球肾炎示意图
1.膜性肾小球肾炎的病变特点示上皮细胞下小丘状电子致密沉积物及毛细血管基底膜形成钉状突起插在沉积物之间2.膜性肾小球肾炎病变发展过程
Ⅰ期:基底膜表面小形丘状沉积物Ⅱ期:基底膜表面形成钉状突起插入沉积物之间,银染色形似梳齿Ⅲ期:沉积物被增生的基底膜包围,埋藏于基底膜内Ⅳ期:基底膜增厚,其中部分沉积物消散,呈虫蚀状
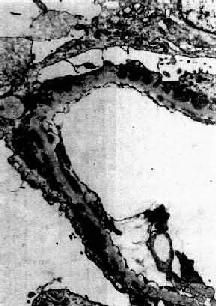
图12-18 膜性肾小球肾炎
电镜下见肾小球毛细血管基底膜表面,上皮细胞下电子致密沉积物,基底膜向表面突出伸入沉积物之间,上皮细胞足突融合
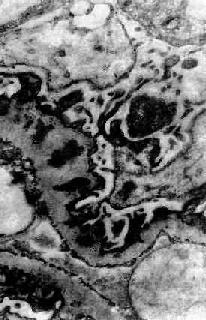
图12-19 膜性肾小球肾炎
电镜下见肾小球毛细血管基底膜增厚,电子致密沉积物被增生的基底膜包围并埋藏于基底膜内
肾小球毛细血管损伤,通透性显著增加,大量蛋白由肾小球滤过进入肾小管,部分被肾小管再吸收。近曲小管上皮细胞浊肿,胞浆内常有玻璃样变颗粒和大量脂肪空泡。晚期随肾小球病变加重,肾小管也萎缩。间质纤维组织增生,炎性细胞浸润较少。
肉眼观,早期可见肾肿胀,体积增大,色苍白。切面皮质明显增宽,髓质无特殊变化。晚期,肾体积缩小,表面呈细颗粒状。
【临床病理联系】
膜性肾小球肾炎是引起肾综合征最常见的原因之一。成人肾病综合征由膜性肾小球肾炎引起者约占40%,小儿仅占5%。膜性肾小球肾炎时,肾小球基底膜严重损伤,通透性显著增加,大量蛋白包括大分子蛋白都可由肾小球滤过引起严重的非选择性蛋白尿。由于大量蛋白由尿中排出,血浆蛋白降低,引起低蛋白血症,血浆胶体渗透压降低,血管内液体渗入组织间隙,引起水肿。同时血容量减少,肾小球血流量和肾小球滤过减少,醛固酮和抗利尿激素分泌增加,引起水钠潴留,进一步加重水肿。因此水肿很严重,往往为全身性,以眼睑和身体下垂部分最明显,严重者并可有胸水和腹水。
高脂血症的原因还不很清楚,可能由于低蛋白血症刺激肝合成各种血浆蛋白包括脂蛋白增多,因此病人有高脂血症和高胆固醇血症。由于血脂过高,血浆内的脂蛋白也可由肾小球滤过,引起脂尿症。
膜性肾小球肾炎时,肾小球内无明显增生和炎症现象。早期,毛细血管不狭窄,血流通畅,故血尿不多见,血压不高,无明显氮质血症。晚期,毛细血管阻塞,肾小球硬化,可引起高血压和肾功能衰竭。
【结局】
膜性肾小球肾炎起病缓慢,病程较长。病变轻者,症状可消退或部分缓解。多数则反复发作,对皮质激素治疗效果不显著。发展到晚期,大量肾单位纤维化、硬化,可导致肾功能衰竭和尿毒症。
六、轻微病变性肾小球肾炎(脂性肾病)
轻微病变性肾小球肾炎(minimal change glomerulonephrits)的病变特点是在光学显微镜下肾小球无明显变化或病变轻微,故名。由于本病肾小管上皮细胞内常有大量脂质沉积,故又称为脂性肾病(lipoid nephrosis)。临床上常表现为肾病综合征。脂性肾病多见于小儿,成人患者较少,是引起小儿肾病综合征最常见的原因。在小儿肾病综合征中约65%~85%由脂性肾病引起。成人肾病综合征由脂性肾病引起者约占10%~30%。脂性肾病病变可完全恢复,对皮质激素治疗效果很好。
轻微病变性肾炎与其他类型肾炎不同,电镜观察未找到沉积物,免疫荧光检查也未发现肾小球内有免疫球蛋白或补体。其病因和发病机制尚不清楚。有些现象如有些病人在呼吸道感染后发病,或同时伴有其他过敏性疾病,如过敏性鼻炎和湿疹等,提示可能与免疫机制有关。Hodgkin病患者轻微病变性肾炎发病率较高,而已知Hodgkin病患者常有T细胞功能缺陷,故有人认为这种肾炎的发病可能与T细胞功能异常有关。近来有学者提出,肾小球多聚阴离子减少可能是造成本病肾小球毛细血管上皮细胞变化和大量蛋白尿的原因。
【病理变化】
光学显微镜下,肾小球无明显变化或仅有轻度节段性系膜增生。电镜下见弥漫性肾小球脏层上皮细胞足突消失(图12-20),细胞内高尔基器和内质网增多,并可见脂滴。细胞表面常有多数微绒毛形成。足突消失不仅见于脂性肾病,也常见于其他原因引起的大量蛋白尿和肾病综合征。经过治疗或蛋白尿等症状缓解后,脏层上皮细胞的变化可恢复正常。
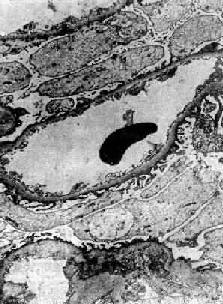
图12-20 轻微病变性肾小球肾炎
电镜下见肾小球毛细血管上皮细胞部分足突消失
肾小管上皮细胞内有多数玻璃样小滴和脂类沉积,是由于肾小球毛细血管通透性增加,大量脂蛋白通过肾小球滤出,而在肾小管被重吸收所致。肾小管腔内可有透明管型。这些变化常与蛋白尿的程度平行。
肉眼观,肾肿胀,体积较大,色苍白。由于大量脂类沉着,切面可见黄色条纹。
【临床病理联系】
脂性肾病病人临床上大多表现为肾病综合征,有大量蛋白尿和严重水肿,与膜性肾小球肾炎不同的是,蛋白尿为高度选择性,主要含小分子蛋白,如白蛋白。肾小球的病变轻微,故一般无血尿和高血压,肾功能也不受影响。
【结局】
大多数病人对皮质激素治疗效果很好,90%以上儿童可以完全恢复,病变在数周内消失。成人预后也很好。少数病人可有反复,一般不发展成慢性。
七、局灶性节段性肾小球硬化
局灶性节段性肾小球硬化(focal segmental glomeralosclerosis)病变为局灶性,节段性,仅累及少数或部分肾小球。病变的肾小球毛细血管丛呈节段性硬化,是引起肾病综合征常见的原因之一。多见于儿童和青年人。此型肾炎病变为进行性,病人对激素治疗效果不理想。多继续发展为弥漫性硬化性肾小球肾炎。
【病理变化】
病变为局灶性,往往从肾皮质深部近髓质部分的少数肾小球开始。早期仅少数肾小球受累,其他肾小球无明显病变或病变轻微。病变的肾小球毛细血管丛的部分毛细血管萎陷,系膜增宽、硬化、玻璃样变(图12-21)。系膜内和毛细血管内常有脂滴和玻璃样物质沉积。有时可见吞噬脂类的泡沫细胞聚积。电镜下可见硬化部分毛细血管基底膜皱缩,厚薄不均匀。其间可见电子致密物和脂滴沉积。上皮细胞足突消失。免疫荧光检查可见病变肾小球内有免疫球蛋白和补体沉积,主要为IgM和C3。
图12-21 局灶性节段性肾小球硬化
肾小球毛细血管丛的一段硬化,玻璃样变
病变继续发展,受累的肾小球逐渐增多。有些肾小球毛细血管丛可全部纤维化、硬化、玻璃样变(球性硬化)。相应的肾小管也萎缩、纤维化。肾间质纤维组织增生,有少量淋巴细胞和单核细胞浸润,有时并可见泡沫细胞聚积。晚期,大量肾小球硬化可发展为弥漫性硬化性肾小球肾炎而导致肾功能不全。
【临床病理联系和结局】
局灶性节段性肾小球硬化病人约80%表现为肾病综合征。但与一般肾病综合征不同,其中约2/3同时伴有血尿,并常有高血压。大量蛋白尿,多为非选择性。病人对激素治疗效果不好,有效率仅为20%~30%。病变为进行性,常继续发展,可导致肾功能不全。一般儿童预后比成年人好。
八、局灶性节段性肾小球肾炎
局灶性节段性肾小球肾炎(focal segmental glomerulonephritis)的病变特点为局灶性和节段性,仅累及部分肾小球,而受累肾小球的病变仅限于肾小球毛细血管丛的一部分。局灶性节段性肾小球肾炎可继发于全身性疾病如系统性红斑狼疮、过敏性紫癜、结节性多动脉炎和细菌性心内膜炎等。有些为原发性,原因不明。
【病理变化】
病变主要为毛细血管丛呈节段性增生,病变轻者可自行消退,有些病变严重毛细血管袢可发生节段性坏死、破裂、出血,有纤维蛋白沉积,并可引起血栓形成。肾球囊内的红细胞和纤维蛋白可引起新月体形成。病变部分修复愈合,纤维化后形成节段性瘢痕。
【临床病理联系和结局】
局灶性节段性肾小球肾炎最常见的症状为镜下或肉眼血尿,有时可出现蛋白尿,偶然可出现肾病综合征,但一般程度较轻。由于病变为局灶性、节段性,故对肾功能影响较小。多数可自行消退,或修复痊愈。有些病人有复发倾向。少数病变广泛者可发展为弥漫性硬化性肾小球肾炎,引起肾功能不全。继发性局灶性节段性肾小球肾炎的预后与原发疾病有关。
九、IgA肾病
IgA肾病(IgA nephropathy,Berger病)是一种特殊类型的肾小球肾炎,由Berger于1968年首先报告。这种疾病的特点为肾小球系膜增生,用免疫荧光法检查见系膜内有IgA沉积。病人的主要症状为复发性血尿,可同时伴有轻度蛋白尿,少数病人可出现肾病综合征。IgA肾病比较常见,多发生于儿童和青年人,男性多于女性,有些病人在发病前有上呼吸道感染史。
【病理变化】
病变程度差异很大。早期病变轻微,呈局灶性,仅少数肾小球有轻度系膜增宽和节段性增生。有些病变较明显,可有弥漫性系膜增生,偶有少数可发展为新月体性肾小球肾炎。最突出的特点是在系膜区内有IgA沉积,常同时并有C3,IgG和IgM较少。早期的补体成分C1q和C4很少见。电镜观察证实系膜中有电子致密物沉积。
过去认为IgA肾病预后很好,近来经过长期随访观察发现IgA肾病多呈慢性进行性过程。约半数病人病变逐渐发展,可出现慢性肾功能不全。
十、弥漫性硬化性肾小球肾炎
弥漫性硬化性肾小球肾炎(diffuse sclerosing glomerulonephritis)是各种类型肾小球肾炎发展到晚期的结果。大量肾小球纤维化、硬化,原始的病变类型已不能辨认。病人的主要症状为慢性肾功能衰竭。有些病人过去有肾炎的病史。约25%的病人起病缓慢,无自觉症状,无肾炎病史,发现时已为晚期。
【病理变化】
两侧肾对称性萎缩变小,色苍白,表面呈弥漫性细颗粒状。有时可有散在的小囊肿形成。肾不而质硬,故称颗粒性固缩肾。切面见肾皮质萎缩变薄,纹理模糊不清,皮髓质分界不明显。肾盂周围脂肪组织增多。小动脉壁增厚,变硬,口哆开。
镜下可见大量肾小球纤维化及玻璃样变,有的形成无结构的玻璃样小团(图12-22)。这些肾小球所属的肾小管也萎缩、纤维化、消失。纤维组织收缩,使纤维化、玻璃样变的肾小球相互靠近集中(图12-23)。有些纤维化的肾小球消失于增生的纤维结缔组织中。无法辨别原有的病变类型。存留的肾单位常发生代偿性肥大,肾小球体积增大,肾小管扩张。肾小管上皮细胞呈立方或高柱状,有些肾小管明显扩大呈小囊状,上皮细胞扁平。扩张的肾小管腔内常有各种管型。间质纤维组织明显增生,并有多数淋巴细胞和浆细胞浸润。由于肾组织缺血及高血压,间质内小动脉硬化,管壁增厚,管腔狭小。
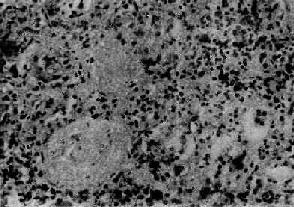
图12-22弥漫性硬化性肾小球肾炎
肾小球纤维化,玻璃样变;肾小管也大部分萎缩纤维化;间质纤维组织增生,有多数慢性炎细胞浸润

图12-23弥漫性硬化性肾小球肾炎
大量肾小球纤维化,玻璃样变,相互靠近、集中,残存的肾小球呈代偿性肥大
【临床病理联系】
晚期硬化性肾炎病人常有贫血、持续性高血压和肾功能不全,而尿常规检查往往变化不明显。由于大量肾单位被破坏,功能丧失,存留的肾单位相对比较正常,血浆蛋白漏出不多,因而蛋白尿、血尿、管型尿都不如早期那样明显,水肿也很轻微。大量肾单位丧失后,血流只能通过存留的肾单位,故血流通过肾小球的速度加快,肾小球滤过率和尿液通过肾小管的速度也随之加快。但肾小管的重吸收功能有一定限度,所以大量水分不能再吸收,肾的尿浓缩功能降低,从而出现多尿、夜尿,尿的比重降低,常固定在1.010左右。
晚期,大量肾单位纤维化,肾组织严重缺血,肾素分泌增加,病人往往有明显的高血压。高血压可促使动脉硬化,进一步加重肾缺血,使血压持续在较高水平。长期高血压可引起左心室肥大,严重时甚至可导致心力衰竭。
晚期肾炎时肾单位大量破坏,残留的肾单位逐渐减少,最后造成体内代谢废物不能排出,水电解质代谢和酸碱平衡调节发生障碍。最后可导致氮质血症和肾功能衰竭。
此外,由于肾组织大量破坏,促红细胞生成素生成减少,长期肾功能不全引起的氮质血症和自身中毒抑制骨髓造血功能,故病人常有贫血。
【结局】
有些肾炎病变发展缓慢,病程长短不一,可达数年至数十年之久。早期进行合理治疗控制疾病发展,可取得较好的效果。病变发展到晚期,大量肾单位被破坏,如不采取措施可导致肾功能衰竭和心功能衰竭,故应注意预防和早期治疗。
硬化性肾炎的死亡原因主要为肾功能不全引起的尿毒症。其次为高血压引起的心力衰竭和脑出血,以及机体抵抗力降低而引起的继发感染。
第二节 尿毒症
肾功能严重障碍,代谢废物不能排出体外,以致大量含氮代谢产物及其他毒性物质在体内蓄积,水电解质代谢及酸碱平衡紊乱,机体内环境的相对稳定被破坏,由此所引起的自身中毒和产生的综合病征称为尿毒症(uremia)。常表现为氮质血症,血尿素氮和肌酐显着升高,并伴有胃肠、神经肌肉和心血管系统的症状,如恶心、呕吐、腹泻、头痛、无力、淡漠、失眠、抽搐、嗜睡以至昏迷等症象。
引起尿毒症的原因很多。肾本身的疾病,如慢性肾小球肾炎,肾盂肾炎等;全身性疾病引起的肾疾病如高血压性肾硬化,系统性红斑狼疮等;以及尿路阻塞等引起肾实质严重损伤,大量肾单位破坏,造成严重肾功能障碍时都可出现尿毒症。
【病因和发病机制】
尿毒症时含氮代谢产物和其他毒性物质不能排出乃在体内蓄积,除造成水、电解质和酸碱平衡紊乱外,并可引起多个器官和系统的病变。
1.消化系统体内堆积的尿素排入消化道,在肠内经细菌尿素酶的作用形成氨,可刺激胃肠粘膜引起纤维素性炎症,甚至形成溃疡和出血。病变范围广,从口腔、食管直至直肠都可受累。以尿毒性食管炎、胃炎和结肠炎较为常见。病人常有恶心、呕吐、腹痛、腹泻、便血等症状。
2.心、肺病变水钠潴留、肾缺血、肾素分泌增加引起的高血压长期作用于心可引起心力衰竭。血液内尿素过高渗入心包和胸膜可引起纤维素性心包炎和纤维素性胸膜炎,听诊时可听到心包和胸膜摩擦音。心力衰竭可引起肺水肿。血尿素从呼吸道排出可引起呼吸道炎症,有时沿肺泡壁可有透明膜形成;肺毛细血管通透性增加,肺泡腔内有大量纤维蛋白及单核细胞渗出,很少中性粒细胞,称为尿毒症性肺炎。
3.造血系统主要改变为贫血和出血。贫血原因:①严重肾组织损害时促红细胞生成素产生不足。②体内蓄积的代谢产物,有些如酚及其衍生物可抑制骨髓的造血功能。另一些毒物如胍及其衍生物可缩短红细胞生存期,加速红细胞破坏并可引起溶血。③转铁蛋白从尿中丧失过多,造成体内铁的运输障碍。
尿毒症病人常有出血倾向,表现为牙龈出血、鼻衄、消化道出血等。出血的原因:①毒性物质抑制骨髓,血小板生成减少。②有些病人血小板数量并不减少,却有出血倾向。这可能是由于血液内胍类毒性物质造成血小板功能障碍,使血小板凝聚力减弱和释放血小板第3因子的作用降低所致。
4.骨骼系统尿素症时常有低血钙。这可能由于:①肾排泄磷酸盐功能下降,故血中磷酸盐浓度升高,钙浓度下降。②体内蓄积的磷酸盐在肠内与食入的钙结合成不溶解的磷酸钙,使钙吸收减少,排出增多。③1,25-二羟胆钙化醇是维生素D在肠道内促进钙吸收的活动形式,在肾内合成。慢性肾疾病时,1,25-二羟胆钙化醇合成发生障碍,致小肠的钙吸收不良,引起低血钙。
长期尿毒症时血钙减少可引起甲状旁腺功能亢进,因而引起骨组织普遍脱钙,称为肾性骨营养不良(renal osteodystrophy),其形态与骨软化(osteomalacia)和囊状纤维性骨炎(osteitis fibrosa cystica)相似。临床上使用1,25-二羟胆钙化醇及其类似药物治疗这些与肾疾病有关的钙代谢障碍效果很好。
5.皮肤尿毒症病人皮肤常呈灰黄色并有瘙痒,皮肤的颜色与贫血和尿色素(urochrome)在皮肤内积聚有关。体内蓄积的尿素可通过汗腺排出,在皮肤表面形成结晶状粉末称为尿素霜,常见于面部、鼻、颊等处。瘙痒的原因不清楚,可能与尿素对神经末梢的刺激有关。
6.神经系统脑组织中大量尿素沉积,渗透压增高,可引起脑水肿,有时有点状出血和小软化灶。毒性物质并可损伤神细胞引起神经细胞变性,血管通透性增高加重脑水肿。尿毒症晚期病人可出现昏睡、抽搐、木僵、昏迷等症状。
有些病人可有周围神经的症状和感觉异常、四肢麻木、烧灼感等。其发生原因还不清楚,可能与甲基胍的含量增高有关。
引起尿毒症的毒性物质尚未完全明了。虽然血尿素含量多少可反映尿毒症的严重程度,但除尿素外,其他代谢产物和毒性物质如胍也有重要作用。尿毒症的发生不是某一种毒素单独作用引起,而是多种因素综合作用的结果。
第三节 肾盂肾炎
肾盂肾炎(pyelonephritis)是一种常见的以肾小管、肾盂和肾间质为主的炎症,可发生于任何年龄,多见于女性,发病率为男性的9~10倍。临床上常有发热、腰部酸痛、血尿和脓尿等症状。根据发病情况和病理变化可分为急性肾盂肾炎和慢性肾盂肾炎两种。
【病因和发病机制】
肾盂肾炎主要由细菌感染引起,肾组织和尿液中都可培养出致病菌。引起肾盂肾炎的致病菌主要为革兰阴性菌,多数为大肠杆菌,约占60%~80%;其次为变形杆菌,产气杆菌,肠杆菌和葡萄球菌等。也可由其他细菌或霉菌引起。急性肾盂肾炎常为单一的细菌感染,慢性肾盂肾炎多为两种以上细菌和混合感染。
肾盂肾炎的感染途径主要有两种:
1.血源性感染细菌由体内某处感染灶侵入血流,随血流到达肾。这种肾盂肾炎可以是全身脓毒血症的一部分。病原菌以葡萄球菌为多见,两侧肾可同时受累。
2.上行性感染下泌尿道的炎症,如尿道炎或膀胱炎时,细菌可沿输尿管或输尿管周围的淋巴管上行到肾盂,引起肾盂和肾组织的炎症。病原菌以大肠杆菌为主。病变可累及一侧或两侧肾。大多数肾盂肾炎为上行性感染。
血源性感染较少见。一般多发生于有输尿管阻塞,体质虚弱和免疫力低下的病人。上行性感染最多见,细菌引起上行性感染,首先要能在尿道粘膜停留繁殖。大肠肝菌细胞膜表面的ρ-菌毛可通过与尿道上皮细胞表面的受体结合粘附在尿道粘膜表面,停留繁殖,对引起上行性感染起重要作用。
尿路的完全或不完全阻塞、尿流不畅引起尿液潴留,有利于细菌感染、繁殖,对肾盂肾炎的发生有重要作用。正常时,机体具有一定的防御功能,包括膀胱内不断有尿液流动,可将进入膀胱的少量细菌稀释冲刷,使之不易在膀胱内停留生长。此外,膀胱粘膜能产生局部分泌型抗体IgA,有抗菌作用。膀胱粘膜内的白细胞也可吞噬杀灭细菌。因此少量细菌进入膀胱后不能生长,膀胱内的尿液无菌。但尿液又是良好的培养基,当某些因素使机体的防御机能削弱时,细菌得以侵入和生长繁殖,引起肾盂肾炎。因此泌尿道结石、前列腺肥大、妊娠子宫和肿瘤的压迫,尿道炎症和损伤后的瘢痕狭窄,以及肾盂输尿管畸形或发育不全等引起尿路阻塞时,容易发生肾盂肾炎。
导尿、膀胱镜检查和其他尿道手术、器械操作等有时可将细菌带入膀胱,并易损伤尿道粘膜引起感染,诱发肾盂肾炎。尤其是留置导尿管的使用,应注意严格灭菌和掌握使用指征。
膀胱输尿管返流是导致细菌由膀胱进入输尿管和肾盂的重要途径。正常人输尿管斜行穿过膀胱壁,在壁内的斜行部分可起瓣膜作用,当排尿膀胱内压增高时,压迫该部输尿管,可阻止膀胱内的尿液返流。如膀胱三角区发育不良,张力减弱,输尿管在膀胱壁内斜行部分过短,输尿管开口异常等,可发生膀胱输尿管返流。此外,下泌尿道梗阻,膀胱功能紊乱,尿道炎,膀胱炎等也可引起膀胱输尿管返流,使细菌进入输尿管和肾盂,引起感染,返流是细菌由膀胱进入肾组织最常见的途径。
女性尿道短,上行性感染机会较多。此外,妊娠子宫压迫输尿管可引起不完全梗阻;黄体酮可使输尿管张力降低,蠕动减弱,容易引起尿滞留,都可诱发感染。同时,男性前列腺液含有抗菌物质,故女性肾盂肾炎发病率比男性高。慢性消耗性疾病如糖尿病和截瘫等全身抵抗力低下时常并发肾盂肾炎。
一、急性肾盂肾炎
急性肾盂肾炎是细菌感染引起的以肾间质和肾小管为主的急性化脓性炎症。
【病理变化】
主要病变为肾间质的化脓性炎和肾小管坏死。病变分布不规则,可累及一侧或两侧肾。肉眼观,肾肿大、充血,表面散在多数大小不等的脓肿,呈黄色或黄白色,周围有紫红色充血带环绕。严重病例数个小化脓灶可融合成大小不等的较大脓肿,不规则地分布在肾组织各部。髓质内可见黄色条纹向皮质伸展。有些和条纹融合形成小脓肿。肾盂粘膜充血、水肿,可有散在的小出血点,有时粘膜表面并有脓性渗出物覆盖,肾盂腔内可有脓性尿液。
上行性感染引起的急性肾盂肾炎首先引起肾盂炎。镜下可见肾盂粘膜充血、水肿,并有中性粒细胞等炎性细胞浸润。以后炎症沿肾小管及其周围组织扩散。在肾间质内引起大量中性粒细胞浸润,并可形成大小不等的脓肿。肾小管腔内充满脓细胞和细菌,故常有脓尿和蛋白尿。尿培养可找到致病菌。早期肾小球多不受影响,病变严重时大量肾组织坏死可破坏肾小球。
血源性感染的特点是肾组织内有多数散在的小脓肿,病变可首先累及肾小球或肾小管周围的间质,并可逐渐扩大,破坏邻近组织,也可破入肾小管蔓延到肾盂。
【合并症】
1.急性坏死性乳头炎主要发生于糖尿病或有尿路阻塞的病人。病变可为单侧或双侧。肉眼观可见肾切面乳头部坏死,范围大小不等。坏死区呈灰黄色,周围有充血带与邻近组织分界明显。镜下见坏死区为缺血性凝固性坏死,坏死区内可见肾小管轮廓,有时可见细菌集落,周围有充血和血细胞浸润。
2.肾盂积脓有严重尿路阻塞,特别是高位完全性尿路阻塞时,脓性渗出物不能排出,淤积充满在肾盂、肾盏和输尿管内,引起肾盂积脓。严重者肾组织受压萎缩变薄,整个肾可变成一个充满脓液的囊。
3.肾周围脓肿肾组织内的化脓性炎症可穿过肾包膜扩展到肾周围的组织中,引起肾周围脓肿。
肾盂肾炎急性期后,肾组织内的中性粒细胞浸润由单核细胞、巨噬细胞、淋巴细胞及浆细胞取代。被破坏的肾组织由纤维组织修补,形成瘢痕,其中可见萎缩的肾小管和多数淋巴细胞浸润。肾盂、肾盏因瘢痕收缩而变形。
【临床病理联系】
急性肾盂肾炎起病急,突然出现发热、寒战、白细胞增多等全身症状。肾肿大和化脓性病变常引起腰部酸痛和尿的变化,如脓尿、蛋白尿、管型尿、菌尿,有时还有血尿等。由于膀胱和尿道急性炎症的刺激可出现尿频、尿急、尿痛等症状。肾盂肾炎病变为不规则灶性,早期肾小球往往无明显病变或病变较轻,故一般肾功能无明显变化,无氮质血症和高血压。
急性坏死性乳头炎时常有明显血尿。严重时肾小管破坏,相应的肾小球被阻塞可引起少尿和氮质血症。乳头坏死组织脱落可阻塞肾盂,有时坏死组织碎块通过输尿管排出可引起绞痛。
【结局】
急性肾盂肾炎如能及时彻底治疗,大多数可以治愈:如治疗不彻底或尿路阻塞未消除,则易反复发作而转为慢性。如有严重尿路阻塞,可引起肾盂积水或肾盂积脓。
二、慢性肾盂肾炎
慢性肾盂肾炎可由急性肾盂肾炎未及时彻底治疗转变而来,或因尿路梗阻未解除,或由于膀胱输尿管返流,病变迁延,反复发作而转为慢性。有些慢性肾盂肾炎病人,多次尿培养皆为阴性,但肾病变反复发作,迁延不愈,可能与免疫反应有关。有人报告,慢性肾盂肾炎病人肾组织中有细菌抗原持续存在,可在体内引起免疫反应,使炎症继续发展。此外,细菌的L型(原生质体)可在肾髓质高渗环境中长期生存,青霉素等许多抗菌药物多系作用于细菌的细胞壁,故对细菌L型无效。细菌L型长期存在与肾盂肾炎发展为慢性有一定关系。
【病理变化】
慢性肾盂肾炎的病变特点是肾小管和肾间质活动性炎症,肾组织纤维化瘢痕形成,肾盂、肾盏变形。病变可累及一侧或两侧肾。病变分布不均匀,呈不规则的灶性或片状。肉眼观,可见两侧肾不对称,大小不等,体积缩小,质地变硬。表面高低不平,有不规则的凹陷性瘢痕(图12-24)。切面可见皮髓质界限模糊,肾乳头部萎缩。肾盂、肾盏因瘢痕收缩而变形。肾盂粘膜增厚、粗糙。

图12-24 慢性肾盂肾炎
两侧肾不对称,体积皆缩小,表面颗粒状,有不规则的瘢痕
镜下见病变呈不规则片状,夹杂于相对正常的肾组织之间。瘢痕区的肾组织破坏,肾间质和肾盂粘膜纤维组织大量增生,其中有大量淋巴细胞、浆细胞、单核细胞和多少不等的中性粒细胞浸润。其间的小血管常有炎症,管壁增厚,管腔狭小。肾小管多萎缩、坏死由纤维组织替代。有些肾小管腔扩张,腔内有均匀红染的胶样管型,形似甲状腺滤泡。有些肾小管腔内还有多数中性粒细胞。肾小球多萎缩、纤维化或玻璃样变(图12-25)。病灶周围的肾组织内肾小球尚完好,有些肾小球的球囊壁增厚,纤维化。有些肾单位呈代偿性肥大。
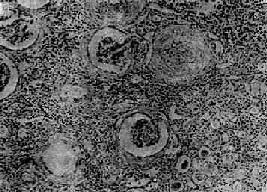
图12-25 慢性肾盂肾炎
有些肾小球毛细血管丛相对正常,肾球囊壁增厚,纤维化,部分肾小管萎缩消失,部分肾小管扩张,腔内有胶样管型;间质纤维组织增生,有大量淋巴细胞等炎细胞浸润
【临床病理联系】
慢性肾盂肾炎常反复急性发作。发作时症状与急性肾盂肾炎相似,尿中有多数白细胞、蛋白质和管型。由于肾小球损害发生较晚,肾小管病变比较严重,发生也较早,故肾小管功能障碍出现较早,也较明显。肾小管浓缩功能降低,可出现多尿和夜尿。电解质如钠、钾等和重碳酸盐丧失过多,可导致缺钠、缺钾和酸中毒。较晚期由于肾组织纤维化和小血管硬化,肾组织缺血,肾素分泌增加,通过肾素-血管紧张素的作用引起高血压。肾乳头萎缩,肾盂肾盏因瘢痕收缩而变形,可通过肾盂造影检见,对临床诊断有一定意义。晚期大量肾组织破坏,可引起氮质血症和尿毒症。
【结局】
慢性肾盂肾炎病程较长,及时治疗,可控制病变发展,肾功能可以得到代偿,不致引起严重后果。若病变广泛并累及双肾者,晚期可引起高血压和肾功能衰竭等严重后果,因此去除诱因和早期彻底治疗非常重要。
第四节 尿石症
尿石症(urolithiasis)是泌尿系统的常见病,多发生于青壮年。尿结石可发生于泌尿道各个部位,如肾盏、肾盂、输尿管、膀胱、尿道等处,是造成尿路阻塞的重要原因之一。
结石的成分根据其形成的原因而不同。但各种结石都包括无机盐结晶和胶体性基质两部分。结晶体约占结石干重的97%~98%,主要有磷酸盐、草酸盐、尿酸盐等。胶体性基质约占结石干重的2%~3%,主要是粘蛋白。在尿结石中含钙者占90%,其中2/3为草酸盐结石,其次为磷酸钙结石和磷酸铵镁结石。不含钙的结石主要有尿酸盐结石(占5%~10%)和胱氨酸结石(占2%~3%)两种。
【结石的类型】
结石的类型取决于其晶体成分。主要有以下几种:
1.草酸盐结石棕褐色,质紧硬,表面粗糙有刺,呈桑椹形,切面呈环形层状。容易损伤尿路粘膜引起血尿,在碱性尿内形成,可以是单纯的草酸钙结石,但多数为草酸钙和磷酸钙混合性结石。
2.磷酸铵镁结石灰白色,表面光滑或有颗粒,质硬或松脆易碎。在肾盂、肾盏内可形成鹿角形结石。切面常见有核心(为细菌或脱落上皮等),呈同心性层状结构。在碱性尿中形成,常与碳酸盐混合。
3.尿酸盐结石黄色或褐色,表面光滑,质硬,圆或卵圆形,常形成多数小结石,在酸性尿内形成。尿酸结石可为单纯性或与草酸钙、磷酸钙等形成混合结石,单纯尿酸结石X线可透过常不显影,混合结石X线不透过可显影。
4.胱氨酸结石 黄白色、光滑、外观蜡样,X线能透过,不易显影,形成于酸性尿中。
大多数尿结石的晶体成分为混合性,单一成分者较少。
【形成原因】
尿结石形成的原因包括尿中晶体浓度过高和尿液理化性质改变两个方面。
1.尿内晶体浓度增高正常尿中常含有多种晶体盐类,如草酸盐、磷酸盐、碳酸盐、尿酸盐等。这些晶体盐类与尿中的胶质物质如粘蛋白类和核酸维持相对平衡。若晶体盐类浓度增高或粘多糖类发生量或质的异常,乃造成晶体与胶体的平衡失调,晶体物质即可析出沉淀,形成结石。当脱水,尿量减少,尿浓缩时,尿中晶体盐类浓度增高,尿结石的发生率增加。
有些情况使体内晶体排出增多,也可使尿晶体浓度增高。95%以上的尿结石含钙,如甲状旁腺功能亢进时可动员骨钙入血;大量肾上腺皮质激素引起溶骨可使尿钙增高;长期卧床病人发生废用性骨萎缩、骨质疏松、脱钙,钙经血流由肾排出;长期服用大量含钙抗酸药物或过量维生素D使钙吸收增多,也可使尿钙增多。如尿中胶体不能维持钙盐的过饱和状态,则钙盐析出沉淀亦可形成结石。
有些代谢紊乱,如痛风病人嘌呤代谢紊乱尿酸排泄增加,可并发尿酸结石。
2.尿液理化性质的改变尿液内晶体浓度正常,但尿液理化性质改变时,也可促进结石形成。如尿液pH改变可影响晶体的溶解度。碱性尿有利于磷酸钙、磷酸氨镁、草酸钙结石形成;酸性尿内易形成尿酸结石和胱氨酸结石。
此外,构成尿结石基质的粘蛋白在尿内含量的改变对尿结石的形成可能也有一定作用。
尿滞留有利于水分再吸收,使尿液浓缩,晶体易析出,同时容易发生感染,析出的晶体可粘附在细菌表面形成结石。
尿内异物,如脱落的上皮细胞、血凝块、炎性渗出物和细菌等可构成结石的核心,尿中晶体盐类可沉积于其上,形成结石。
【病理变化及对机体的影响】
尿结石形成最常见的部位为肾盂、肾盏和膀胱。约80%的病人为单侧性。
尿结石大小不一,大者直径可达数厘米,小者如砂粒;数量也不等,少者只有一个,多者可有数十个甚至数百个(尿砂),如膀胱内的泥砂样结石。尿结石的形状可为圆形、椭圆或不规则形(如肾盂内的鹿角形结石)。表面的的光滑,有的粗糙。尿结石对机体的影响主要是引起泌尿道阻塞和损伤。表面光滑的结石固定在肾盂内如不移动可不出现症状。结石阻塞肾盂和输尿管可引起肾盂积水和输尿管积水。有些结石可损伤肾盂、输尿管和膀胱粘膜引起血尿。尿结石的慢性刺激可引起粘膜慢性炎症、鳞状化生成白斑。小形尿结石进入输尿管,可招致强烈蠕动和痉挛,引起剧烈的绞痛。嵌顿在输尿管的结石常损伤输尿管粘膜引起溃疡形成,以后可能造成输尿管瘢痕狭窄。由尿结石造成的阻塞和损伤又是诱发感染的重要因素,因此常并发尿路感染和肾盂肾炎。
第五节 肾盂积水
肾盂积水(hydronephrosis)是由于尿路阻塞而引起的肾盂肾盏扩大伴有肾组织萎缩。尿路阻塞可发生于泌尿道的任何部位,可为单侧或双侧。阻塞的程度可为完全性或不完全性,持续一定时间后都可引起肾盂积水。
【尿路阻塞的原因】
尿路阻塞可突然发生,也可逐渐缓慢形成。常见的引起尿路阻塞的原因如下。
1.腔内阻塞①先天性畸形如输尿管或后尿道瓣膜和尿道狭窄等;②异物如尿结石、血凝块、肾乳头坏死脱落等;③肿瘤如肾盂、肾盏、输尿管及膀胱肿瘤;④炎症如输尿管炎和尿道炎等。
2.腔外压迫①膀胱和输尿管邻近组织的肿瘤,如前列腺癌,子宫体及子宫颈癌,后腹膜肿瘤等;②良性前列腺肥大;③泌尿道周围组织的炎症和后腹膜纤维化;④肾下垂伴输尿管扭曲;⑤妊娠子宫。
3.功能紊乱神经性功能紊乱,如脊髓损伤引起膀胱麻痹,其他如输尿管、膀胱及尿道的功能紊乱性阻塞。
【病理变化】
尿路即使完全阻塞后,在一定时期内肾小球滤过仍继续进行。但因肾小球滤液不能排出,故一方面向肾间质及周围组织弥散,最后进入淋巴管或静脉;另方面在肾盏肾盂内蓄积,使肾盂肾盏逐渐扩张,肾盂内压力增高,压迫肾组织及其中的血管,使血流减少,以后肾组织受压逐渐萎缩。早期主要累及肾小管,表现为肾浓缩功能降低;以后逐渐影响肾小球,使肾小球滤过减少。完全性尿路阻塞一般约三周、不完全性阻塞约三个月可造成严重的不可逆性损伤。
肾盂积水可为单侧或双侧。突然发生的完全性尿路阻塞,一般引起肾盂肾盏轻度扩大,有时可引起肾组织萎缩。间断发生的不完全尿路阻塞时,肾盂、肾盏逐渐进行性扩大。根据阻塞部位高低,膀胱、输尿管或肾盂可先后发生扩张。一般低位阻塞时,常两侧肾受累,肾盂肾盏尚未扩张到很严重的程度时尿毒症即可出现。
肉眼观,肾呈不同程度增大。早期仅肾盂肾盏扩大,逐渐肾锥体乳头变钝平,以后呈杯状。晚期肾组织萎缩,皮质变薄,锥体消失。整个肾可成为一个薄壁的囊,直径可达15~20cm。
镜下,早期可见肾小管扩张,肾小球无明显变化,渐而肾小管萎缩、纤维化,肾小球也逐渐萎缩消失,肾组织终为薄层纤维组织所替代。
【临床病理联系】
急性阻塞时肾膨胀牵引肾包膜可引起疼痛。早期症状与引起肾盂积水的原因有关,如结石阻塞输尿管可引起绞痛;前列腺肥大压迫尿道和膀胱可引起排尿困难和膀胱内尿潴留等症状。
一侧性完全或不完全阻塞性肾盂积水时,由于对侧肾功能正常,故多无明显症状而常不被发现,病变常继续发展,最后病侧肾组织可全被破坏。如能早期发现及时解除尿路阻塞,肾功能可能恢复。
两侧性不完全阻塞,首先影响肾小管,主要表现为肾浓缩功能降低,出现多尿、夜尿。有些病人可发生肾小管性酸中毒。两侧性完全阻塞如不解除,可导致少尿、无尿和尿毒症。
第六节 肾肿瘤
一、肾细胞癌
肾细胞癌(renal cell carcinoma)是肾原发肿瘤中最多见的,约占肾恶性肿瘤的80%~90%。多发生于60岁左右的老年人。男性多于女性,约为2:1。肾细胞癌来源于肾小管上皮细胞,故又称为肾腺癌。
【病理变化】
肾细胞癌可发生于肾的任何部位,但多见于肾之两极,尤以上极更为多见。一般为单个圆形,大小差别很大,小者直径1~2cm,大者可重达数公斤,多数直径为3~10cm。切面上癌组织呈淡黄色或灰白色,其间常有出血、坏死、软化和钙化区,故常呈红、黄、灰白相间的多种色彩(图12-26)。癌组织与邻近的肾组织分界明显,常有假包膜形成。但肿瘤周围组织中常可见小肿瘤结节环绕,说明肿瘤具有侵袭性。肿瘤逐渐生长可侵入肾盂、肾盏,引起阻塞,导致肾盂肾盏扩张和肾盂积水。肿瘤并可突破肾盂侵入输尿管。此外,肾细胞癌的特点是常侵入肾静脉,并可在静脉腔内生长呈条索状向下腔静脉延伸,甚至可达右心。有时癌组织可穿破肾包膜,侵犯肾上腺和肾周围软组织。
图12-26 肾细胞癌
肾由大形肿瘤占据,左侧可见残存的肾组织
镜下,癌细胞形态和排列多样,多数癌细胞体积较大,呈多角形,轮廓清楚,胞浆清亮透明;细胞核小而深染,位于细胞中央或边缘。癌细胞常排列成片状(图12-27)、条索状、腺泡状或管状,很像肾小管(图12-28)。以这种形态结构为主的称为透明细胞癌。这种类型在肾细胞癌中最为多见。有些肾细胞癌由颗粒细胞组成。癌细胞体积较大,呈立方形或多角形,胞浆呈伊红色细颗粒状;核圆形,多位于细胞中央。分化较好的癌细胞比较规则,排列成腺状或片状;分化不好的癌细胞大小形状不规则,胞浆深染,核大并有多数核分裂像,有时并有癌巨细胞形成。高度未分化的癌细胞呈梭形或不规则形,似肉瘤样,称为未分化癌。有时癌组织中有大量乳头状结构,这种结构可见于透明细胞癌的某些区域,也可为肾细胞癌的主要成分,称为乳头状腺癌。有时其间并有囊肿形成则称为乳头状囊腺癌。以上各种类型之间常有过渡形式。在大多数肾癌中,几种类型常同时并存,而以某一类型为主。癌组织的间质很少,但血管丰富,有些区域纤维组织可增多,常有出血、坏死和钙化。
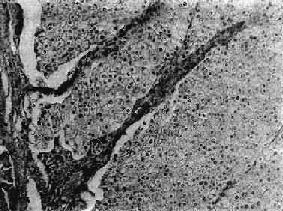
图12-27 肾透明细胞癌
癌细胞呈多角形或立方形,轮廓清楚,胞浆透明,核居中,深染。癌细胞排列成片状,间质很少
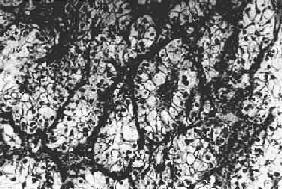
图12-28 肾透明细胞腺癌
癌细胞大,胞浆透明,排列成腺样结构,间质少
【转移】
肾细胞癌除直接蔓延向邻近组织扩散外,可通过血道和淋巴道转移。癌组织血管丰富,早期即可发生血路转移。有时原发灶体积很小,无局部症状,但已侵入肾静脉引起远处转移。最常转移到肺,其次为骨、局部淋巴结、肝、肾上腺和脑。有些肾细胞癌可转移至对侧肾。有些可转移到一些不寻常的部位,如口腔、喉、眼和阴道。左肾静脉内的癌细胞栓可沿精索静脉或卵巢静脉逆行转移到生殖器官。淋巴道转移常首先到肾门及主动脉旁淋巴结。
【临床病理联系】
肾细胞癌早期常无症状,或只有发热、乏力等全身症状,肿瘤体积增大时才被发现。临床主要表现为血尿、肾区痛和肿块。血尿多因癌组织浸润血管或侵及肾盂或肾盏而引起。肿瘤体积大或侵犯包膜时,可引起腰部疼痛,并可触及肿块。有时癌组织出血,血块通过输尿管排出时,可引起剧烈绞痛。
肾癌可产生多种激素和激素样物质而引起各种不同的症状。其中包括①红细胞生成素可引起红细胞增多症;②甲状旁腺样激素可引起血钙过高;③肾素可引起高血压;④促性腺激素可导致女性化或男性化;⑤肾上腺糖皮质激素可引起Cushing综合征。
如无远处转移,则早期彻底手术切除,包括清除肾门、主动脉旁淋巴结,预后较好。若癌细胞侵入肾静脉或侵犯肾周围组织则预后差。
二、肾母细胞瘤
肾母细胞瘤(nephroblastoma,wilms’tumor)是肾胚基来源的肿瘤。主要发生于儿童,是10岁以下儿童最常见的肿瘤之一。最多见于1~4岁的小儿,很少见于成人。
【病理变化】
肾母细胞瘤通常为单侧,约有5%~10%为双侧。肿瘤多巨大呈球形,有时可占据大部分腹腔。肿瘤界限清楚,压迫周围肾组织,好似有包膜。切面色彩多样,与肿瘤的成分有关:部分呈灰白色,质硬;部分粘液样,质软;部分呈鱼肉状;部分可见透明软骨样组织,并常有钙化、大片出血和坏死区(图12-29)。早期,肿瘤一侧可见残留的肾组织,晚期则肾组织可全部被破坏,并可穿破肾包膜侵入肾周围组织。
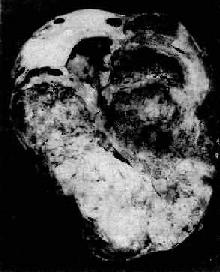
图12-29 肾母细胞瘤
镜下可见原始的肾小球样和肾小管样结构,周围为间叶组织来源的梭形细胞基质(图12-30)。梭形细胞胞浆少,核染色深,呈肉瘤样。此外,肿瘤内还可见横纹肌、平滑肌、胶原纤维、软骨、骨和脂肪细胞。部分有坏死,其中可见胆固醇结晶和吞噬脂类的巨噬细胞。最常见的是横纹肌细胞。基质梭形细胞的异型性程度与预后有关。
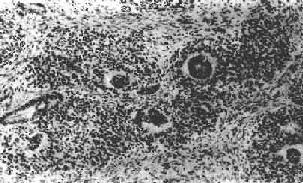
图12-30 肾母细胞瘤
瘤组织内有原始的肾小球和肾小管样结构,周围为梭形细胞基质
【临床病理联系】
腹部肿块是最常见的症状,有些病人可有血尿。巨大的肿块可越过腹中线直到盆腔,压迫邻近器官引起腹痛、肠梗阻。有些病儿有高血压,可能与肿瘤压迫肾动脉和产生肾素有关。
肿瘤在局部生长可侵入邻近组织,也可沿血道和淋巴道转移到肺、肝、肾门淋巴结和主动脉旁淋巴结。综合应用手术切除、放射治疗和化学治疗效果较好。有些已发生肺部转移的病人经综合治疗后,肺部转移灶可消失。
第七节 膀胱肿瘤
膀胱肿瘤中约95%来源于膀胱上皮,少数来源于间叶组织,如纤维组织和肌组织等。膀胱的上皮性肿瘤中绝大多数为恶性,只有少数为良性,包括乳头状瘤、移行细胞癌、鳞状细胞癌和腺癌。其中以移行细胞癌最常见。
一、膀胱移行细胞乳头状瘤
膀胱移行细胞乳头状瘤(transitional cell papilloma)可发生于膀胱粘膜的任何部位,但以膀胱侧壁和三角区最多见。临床上主要表现为无痛性血尿。膀胱乳头状瘤切除后易复发,与低度恶性的乳头状癌难以区别。真正良性的乳头状瘤很少见,在膀胱乳头状肿瘤中仅约占1%~2%。
【病理变化】
乳头状瘤可为单发性或多发性,一般体积较小,约0.5~2.0cm,在膀胱粘膜表面形成乳头状突起。有时乳头细长呈绒毛状或分支状,有蒂与膀胱粘膜相连。细长的乳头漂浮于尿液内,易折断脱落,引起血尿。
镜下可见膀胱粘膜移行上皮增生形成乳头状突起。乳头表面被覆上皮与正常膀胱移行上皮非常相似(图12-31)。细胞大小、排列都很整齐,似正常分化。乳头轴心的间质纤细,由少量纤维结缔组织构成,其中含有少数薄壁毛细血管,并有少量炎性细胞浸润。
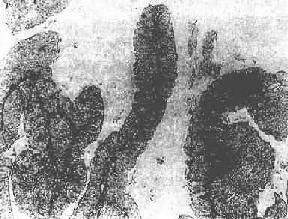
图12-31 膀胱移行细胞乳头状瘤
瘤组织呈乳头状,表面被覆上皮与正常移行上皮相似,细胞大小和排列整齐
膀胱乳头状瘤虽然分化程度高,但手术切除后容易复发,并且随着复发次数增加,分化程度降低,容易发生癌变。因此有人主张把膀胱乳头状瘤看作低度恶性乳头状癌,而称之为移行细胞乳头状癌Ⅰ级。
二、膀胱癌
膀胱癌是泌尿道肿瘤中最多见的。多发生于40~60岁之间,男性比女性多3倍。
【病理变化】
膀胱癌因多发生于膀胱侧壁和三角区近输尿管开口处,故易阻塞输尿管口引起肾盂积水和肾盂肾炎。肿瘤可为单发性或多发性,大小不等,从数毫米至数厘米。外观呈乳头状或扁平。乳头状癌在膀胱粘膜表面形成乳头状突起,有蒂与膀胱粘膜相连,有时呈息肉状或菜花状。分化不好,恶性程度较高的肿瘤多无蒂,基底宽,突出于粘膜表面,并向壁内作不同程度的浸润。有些肿瘤不形成突起,表现为膀胱粘膜局部增厚呈扁平斑块状。这种类型早期可局限于粘膜内,但多数浸润至粘膜下,其恶性程度往往比乳头状癌高,表现可有溃疡形成、出血和伴发感染。
根据组织学类型可将膀胱癌分为移行细胞癌、鳞状细胞癌和腺癌,有些为混合性。其中以移行细胞癌为最常见,腺癌很少见。
1.移行细胞癌(transitional cell carcinoma) 约占膀胱癌的90%,分化程度不同,包括从分化良好的乳头状非浸润性癌到高度未分化的浸润性癌。其中约70%为分化良好的乳头状癌,25%~30%为分化程度不同的浸润性癌。根据癌细胞分化程度不同,将移行细胞癌分为三级。
(1)移行细胞癌Ⅰ级:癌组织呈乳头状,乳头表面被覆的移行上皮较厚,细胞层次较多,缺乏从底层到表层由柱状细胞到扁平细胞逐渐分化的现象。细胞核大小不甚一致,有些较大染色较深。核分裂像可见,有的局部区域稍多,且不限于基底层(图12-32)。有些癌细胞可浸润固有膜。
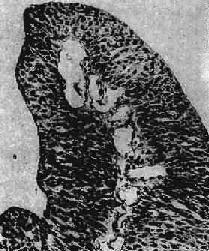
图12-32 膀胱移行细胞癌 Ⅰ级
肿瘤乳头状,被覆上皮细胞层次增多,部分排列不整齐,核大小不甚一致
(2)移行细胞癌Ⅱ级:肿瘤呈乳头状、菜花状或扁平无蒂,表面常有坏死和溃疡形成。镜下,部分癌组织仍保持乳头状结构,但多不规则,并有许多实体癌巢。癌细胞大小不一,排列紊乱,极性消失,常有癌巨细胞形成。核大小不等,染色深,核分裂像较多(图12-33)。癌组织常浸润至上皮下组织,甚至可达肌层。

图12-33 膀胱移行细胞癌 Ⅱ级
癌组织呈乳头状,细胞层次显著增多,大小、形状不一,排列紊乱,核分裂像较多
(3)移行细胞癌Ⅲ级:部分为菜花状,底宽无蒂,或为扁平的斑块,表面常有坏死和溃疡形成。癌细胞高度未化化;细胞大小、形态不一,排列紊乱(图12-34)。很少或无乳头状结构,有的形成不规则的癌巢,有的分散。常有多数瘤巨细胞。核形状不规则,染色深,核分裂像很多,并有多数不典型的病理性核分裂像。癌组织常浸润到膀胱壁肌层深部,并可穿过膀胱壁浸润到邻近器官,如前列腺、精囊、子宫和腹膜后组织等。
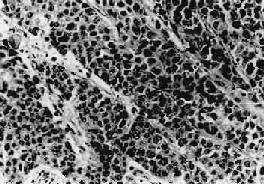
图12-34 膀胱移行细胞癌 Ⅲ级
癌细胞分化不良,失去移行上皮的特征,癌细胞大小、形状不规则,排列紊乱,核分裂像多见
各级膀胱移行细胞癌以及乳头状瘤都有复发倾向,并且复发的肿瘤分化往往更不成熟。有些分化不好的移行细胞癌部分可有鳞状化生,生长较快,预后较差。
2.鳞状细胞癌(squamous cell carcinoma) 较少见,约占膀胱癌的5%,常在膀胱移行上皮鳞状化生的基础上发生。许多病人有慢性炎症合并粘膜白斑。鳞状细胞癌有时可形成肿块突出表面,多数为浸润性,表面常有坏死和溃疡形成。镜下结构与一般鳞状细胞癌相同。分化程度不一,有些分化好的,可见细胞间桥和角化,并可有多数癌珠形成;有些分化差,表现为未分化癌。这种单纯的鳞状细胞癌应与上述移行细胞癌伴有灶性鳞状化生相区别,因为单纯的鳞状细胞癌预后较好。
3.腺癌(adenocarcinoma) 很少见,占膀胱癌的1~2%。膀胱腺癌可来自脐尿管残余、尿道周围和前列腺周围的腺体、囊性和腺性膀胱炎或移行上皮的化生。有些腺癌可产生粘液。这种肿瘤可向粘膜表面突出,发生坏死和溃疡,并可向深部浸润膀胱壁,有些肿瘤表面可有大量粘液覆盖。
【转移】
膀胱癌主要通过淋巴道转移到局部淋巴结,并常侵犯子宫旁、髂动脉旁和主动脉旁淋巴结。晚期可发生血路转移,多发生于高度未分化癌,有些可发生广泛转移,常见于肝、肺、骨髓、肾、肾上腺等处。
【临床病理联系】
各种膀胱肿瘤最常见的症状为无痛性血尿。乳头状癌的乳头断裂、肿瘤表面坏死、溃疡形成以及并发膀胱炎等皆可引起血尿。肿瘤侵犯膀胱壁,刺激膀胱粘膜及并发感染时可引起尿频、尿急和疼痛,多见于Ⅱ级和Ⅲ级膀胱癌。肿瘤如阻塞输尿管开口可引起肾盂肾炎、肾盂积水,甚肾盂积脓。
膀胱肿瘤的预后与肿瘤的分化程度和浸润范围有密切关系,分化程度越高预后越好。乳头状瘤预后很好,单纯切除后,五年生存率可达90%以上。Ⅱ级和Ⅲ级的膀胱癌预后不好。膀胱顶部和前壁肿瘤预后比膀胱底部肿瘤预后差。晚期病人往往死于广泛转移和输尿管阻塞引起的感染。
泌尿系统的癌肿位于腹腔及盆腔,只有用特殊检查方法才能查见,故早期诊断比较困难。做尿液或膀胱冲洗液的脱落细胞学检查有其优越性,但由于肾内的癌细胞必须脱落到肾盂才能排出,而有些分化好的移行细胞癌的脱落细胞无法与正常移行上皮细胞鉴别,故尿液脱落细胞学检查的阳性率和正确率不很高,主要对诊断肾盂、输尿管和膀胱的分化不成熟的移行细胞癌和鳞状细胞癌有所帮助。近来应用流式细胞仪分析肿瘤细胞DNA含量对膀胱肿瘤的诊断,尤其是预后有很大帮助。很多恶性肿瘤细胞为DNA非整倍体性,并且非整倍体肿瘤的侵袭性或恶性程度往往比形态相似的二倍体肿瘤更高。因此尿液或膀胱冲洗液的流式细胞技术分析DNA含量和脱落细胞形态学观察两者结合应用,对膀胱肿瘤的预后、预测治疗效果和复发情况都可取得更好的效果。
第十三章 女性生殖系统疾病
第一节 外阴疾病
一、尖锐湿疣
尖锐湿疣(condyloma acuminatum)主要是由人乳头状瘤病毒(HPV)6型及11型感染引起的良性疣状物。多数通过性接触传播,常有性伴侣同时患尖锐湿疣的病例。发病年龄高峰在20~30岁。少数病例也可通过其他方式交叉感染,偶见于接触感染的婴儿及青春期前儿童。好发部位为小阴唇、阴蒂、处女膜周围、尿道外口、阴道壁及宫颈等处,肛周及会阴部也可受累。潜伏期可长达数月。常有局部瘙痒。
肉眼观,典型病例呈多个小而尖的乳头,淡红色或灰色,较湿润。有些也可呈斑块状或互相融合成结节菜花状。镜下,上皮增生呈乳头状结构,典型者为细长的尖乳头,表面覆盖鳞状上皮,呈不全角化及轻度角化过度。棘细胞明显增生,伴上皮钉突增厚延长。在棘细胞层中,上部可见多少不等的空泡状细胞(koilocytotic cell),核大而且大小不一,染色质粗,深染,核周空晕,在空晕区内可见胞浆细丝(图13-1)。电镜下往往可检见核内病毒颗粒。目前用免疫组化法检测人乳头状瘤病毒核壳抗原及运用原位杂交技术检测HPV DNA,阳性者有助于确诊。
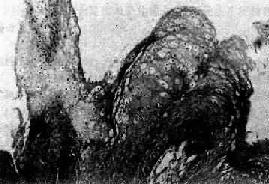
图13-1 外阴尖锐湿疣
上皮增生呈细长尖乳头状,表皮不全角化,棘细胞增生,中表层有许多空泡状细胞
本病临床经过一般慢而长,妊娠期病变发展迅速,分娩后可呈退行性变,也可重复感染及复发。治疗方法有药物、电灼、冷冻及激光疗法等。
近来有些作者提出本病与外阴癌的并发率较高,也有极少数恶变成乳头状原位癌、疣状癌或高分化鳞癌的报道。
二、女阴营养不良
女阴营养不良(vulvar dystrophy)过去称女阴白色病变或白斑,并当作癌前病变,给以不当的处理。实际上外阴营养不良多不属癌前病变,病因未明,主要表现为外阴病损部呈白色,角化过度及色素脱失,伴有不同程度的瘙痒。组织改变可分为增生型、萎缩型及混合型三个基本类型。
1.增生型营养不良(hyperplastic dystrophy)多发生于40~60岁妇女,好发于阴蒂、小阴唇及阴唇沟等处。局部皮肤呈白色,少数呈粉红色,皮肤和粘膜均增厚,粗糙,弹性差,可有皲裂或溃疡形成。镜下,表皮增厚,常有角化过度,棘细胞增生,钉突延长,基底细胞层常见不等量黑色素脱失。真皮乳头水肿,有多少不等的慢性炎性细胞浸润(图13-2)。可伴有或不伴有不典型增生。本病属良性病变,不合并不典型增生者一般不会癌变,故不属癌前病变。用保守疗法和药物治疗可迅速收效。若伴有不典型增生,则有一定恶性潜能,应注意随访病人。

图13-2 增生型女阴营养不良
棘细胞增生伴钉突延长、增宽和融合,表皮角化过度,真皮慢性炎性细胞浸润
2.萎缩性硬化性苔癣(lichen sclerosus et atrophicus)多见于绝经后妇女,好发于阴蒂、小阴唇、会阴和肛周。病变皮肤萎缩变薄、光亮、呈白色。镜下,角质层增厚,可见角栓形成。表皮变薄,上皮脚变短或消失,基底细胞空泡变性及液化,色素脱失。真皮浅层水肿,细胞成分减少,呈带状均质化为其特征,下方常有慢性炎性细胞浸润。萎缩性硬化性苔癣一般不伴有不典型增生,多数不致发展为癌。
3.混合型营养不良(mixed dystrophy)外阴皮损区变白,增生与萎缩病变同时存在。可能伴有不典型增生,故应作多点活检,才能确诊。
第二节 子宫颈疾病
一、慢性子宫颈炎
慢性子宫颈炎(chronic cervicitis)是育龄妇女最常见的疾病。常由链球菌、葡萄球菌及肠球菌引起。临床上主要表现为白带过多。阴道镜可见子宫颈粘膜充血。镜下为子宫颈非特殊性炎症,可见子宫颈内膜上皮下有淋巴细胞、浆细胞及单核细胞浸润。子宫颈柱状上皮及腺上皮常伴有不同程度鳞状上皮化生。少数也可因感染病毒、结核、寄生虫及放线菌等引起特殊性炎症。
慢性子宫颈炎时子宫颈阴道部鳞状上皮有时坏死脱落,形成表浅的缺损,称真性糜烂,较少见。临床上常见的宫颈糜烂(cervical erosion)实质上是宫颈先前损伤的上皮已被宫颈管内柱状上皮外移取代了子宫颈阴道部的鳞状上皮。由于单层柱状上皮很薄,使上皮下血管容易显露而呈红色,看上去像糜烂,实际上为假性糜烂。多发生在育龄或卵巢功能旺盛的妇女,由于雌激素水平增高,使宫颈管内柱状上皮增生越出宫颈外口。间质内常无明显炎症现象,在产后或绝经后可自行消退。
慢性子宫颈炎时子宫颈腺的颈部易被增生的纤维组织所压迫,或由于腺腔被粘液或化生的鳞状上皮阻塞,使粘液潴留,腺体扩大成囊状,直径一般在数毫米至1厘米,称子宫颈腺囊肿,又称纳博特囊肿(Nabothian cyst)。
有些病例由于子宫颈粘膜、腺体和固有膜结缔组织呈局限性增生而形成子宫颈息肉(cervical ployp)。直径自数毫米到数厘米不等。呈粉白色或粉红色,常有蒂。镜下主要由腺体和结缔组织构成。间质充血、水肿及多少不等的慢性炎性细胞浸润。表面被覆单层柱状上皮或鳞状上皮。宫颈息肉属良性瘤样病变,恶变率很低,在1%以下。
二、子宫颈上皮不典型增生
子宫颈上皮不典型增生(dysplasia of cervical epithelium)属癌前病变。表现在子宫颈上皮层内出现异型细胞:细胞核大、浓染、染色质增粗、核大小不一、形状不规则、核分裂像增多、有病理性核分裂、细胞极性紊乱等。不典型增生可分三级:Ⅰ级(轻度),上述异型细胞局限于上皮层的下1/3区。Ⅱ级(中度),增生的异型细胞占上皮层下部的1/3至2/3。Ⅲ级(重度),增生的异型细胞超过全层的2/3。一般,级别越高,发展为浸润癌的机会越多;级别越低,自然消退的机会也越多。轻度不典型增生在子宫颈慢性炎症中常可见到,多可迅速恢复,其恶变率很低。中度不典型增生可发展为重度不典型增生。而重度不典型增生则具有高度恶变危险。子宫颈不典型增生经过治疗后,多数可消退,有些病例可长期存在,少数病例最终可发展为癌。
三、子宫颈原位癌
子宫颈原位癌(carcinoma in situ)癌细胞局限于上皮全层内、尚未穿破上皮基底膜侵入下方固有膜。子宫颈原位癌的异型细胞比不典型增生者更具显着的多形性。镜下,上皮层完全为癌细胞所取代,细胞大小、形状不一,呈圆形、卵圆形、梭形,偶见巨核、多核,排列紊乱,层次不清,极向消失。核大浓染、大小及形状不一、染色质增粗,核分裂像常见,并有病理性核分裂像。胞浆相对减少,核浆比值增大(图13-3)。原位癌癌细胞可由表面沿基底膜伸入腺体内,致整个腺管或其一部分为癌细胞所取代,但腺管轮廓尚存,腺体基底膜完整,癌细胞未浸润到固有膜。这种变化称原位癌累及腺体。原位癌累及腺体并不一定发展为浸润癌。部分子宫颈原位癌可长期不发生浸润,个别病例甚至可自行消退。但由于原位癌特别是原位癌累及腺体具有发展为浸润癌的倾向,故一旦发现,应及时给予适当治疗。
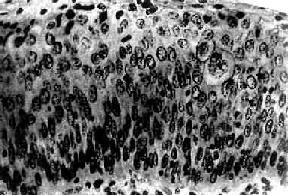
图13-3 子宫颈原位癌
癌变限于上皮层内,细胞核肥大、深染,大小不一,形态不规则,核仁明显,核分裂像易见,且见巨细胞(右侧上方),这种细胞间变累及上皮全层,但基底膜完整,癌细胞未浸润到基底膜下间质
由于重度不典型增生和原位癌没有明显界限,有学者将不同程度的不典型增生和原位癌统称为宫颈上皮内新生物(cervical intraepithelial neoplasm,CIN)。Ⅰ级(轻度)不典型增生为CINⅠ。Ⅱ级(中度)不典型增生为CINⅡ。Ⅲ级(重度)不典型增生及原位癌为CINⅢ。他们认为这些病变是有连续关系的,只是在程度上不同而已。这一组病变是处于正常鳞状上皮和浸润癌之间的变化阶段。上皮的不典型增生-原位癌-浸润癌是一个逐渐连续发展的过程。但并非所有子宫颈浸润癌的形成均必须通过这一过程,也不是所有的上皮不典型增生均必然发展为子宫颈癌。癌前病变具有进展性和可逆性,决定于病变的范围及程度。
四、子宫颈癌
子宫颈癌(carcinoma of cervix)是女性生殖系统中常见的恶性肿瘤之一。发病年龄以40~60岁最多,平均年龄50岁。由于防癌工作的开展,很多子宫颈癌能在早期被发现,因此晚期癌远较过去为少。五年生存率明显提高。目前对宫颈癌的临床和病理工作也都着重于对早期癌的发现。其研究方向也更着重于对亚临床宫颈癌的诊断。
宫颈癌的病因尚无定论,一般认为与早婚、多产、性生活紊乱、宫颈裂伤、包皮垢及感染等因素有关。近年来对病毒病因研究较多,许多作者通过免疫荧光法在宫颈癌细胞中找到HSV-2型抗原,并从宫颈癌活组织中已分离出HSV-2型病毒DNA。也有人用多聚酶链反应(PCR)及原位核酸分子杂交技术在宫颈癌组织中证实有HPv DNA,说明HPV感染,尤其是HPV16、18、31型与宫颈癌的发病有关。但现有资料尚未能证实病毒是直接致癌的。其作用机制有待进一步研究。
子宫颈癌的组织发生来源主要有三,即宫颈阴道部或移行带的鳞状上皮、柱状上皮下的储备细胞及子宫颈管粘膜柱状上皮。
子宫颈癌的组织类型主要有鳞状细胞癌及腺癌两种。
(一)子宫颈鳞癌
子宫颈鳞癌在子宫颈癌中最为常见,其发生率约占子宫颈恶性肿瘤的90%以上。根据癌发展的过程,可分早期浸润癌及浸润癌。
1.早期浸润癌或微浸润癌(microinvasive carcinoma)是指上皮内癌突破基底膜向固有膜浸润,浸润深度不超过基底膜下3~5mm,在固有膜中形成一些不规则的癌细胞条索或小团块。一般肉眼不能判断,只能在显微镜下证明有早期浸润。早期浸润癌可来源于原位癌的进展或由其它上皮异常甚或完全正常的鳞状上皮增生直接发展形成。
2.浸润癌(incasive carcinoma)指癌组织突破基底膜,明显浸润到间质内,浸润深度超过基底膜下5mm,并伴有临床症状者。肉眼观,主要表现为内生浸润型(图13-4)、溃疡状或外生乳头状、菜花状。镜下,按其分化程度可分为三型:①高分化鳞癌,约占20%,癌细胞主要为多角形,似鳞状上皮的棘细胞,有角化及癌珠形成,核分裂像不多,对放射线不敏感。②中分化鳞癌,约占60%,多为大细胞型,癌细胞为椭圆形或大梭形,无明显癌珠,核分裂像和细胞异型性较明显,对放射线较敏感。③低分化鳞癌,约占20%,多为小细胞型,细胞呈小梭形,似基底细胞,异型性及核分裂像都很明显,对放射线最敏感,但预后较差。
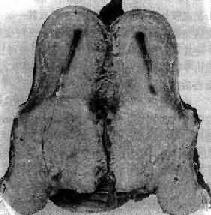
图13-4 子宫颈癌(内生浸润型)
切面见癌组织灰白色,呈结节状在子宫颈管内浸润生长
(二)子宫颈腺癌
子宫颈腺癌较鳞癌少见,其发生率约占宫颈浸润癌的5%左右,近年来报道宫颈腺癌的发病率有上升趋势,约占宫颈浸润癌的8%~12.7%,平均发病年龄56岁,较鳞癌患者的平均年龄大5岁。在20岁以下青年女性的宫颈癌中,则以腺癌为多。有人认为口服避孕药与宫颈腺癌发病率升高有关,但尚不能定论。其组织发生主要来源于宫颈表面及腺体的柱状上皮,少数起源于柱状上皮下的储备细胞。大体类型与鳞癌基本相同。镜下,呈一般腺癌的结构。有些病例表面为高分化类型,往往需多次活检才能证实。有时可表现为乳头状腺癌、透明细胞癌、棘腺癌和腺鳞癌等。宫颈腺癌对放射线不敏感,易早期发生转移,应尽早争取手术治疗,预后较宫颈鳞癌差。
子宫颈癌的扩展和转移
子宫颈癌的主要扩展途径为直接蔓延和经淋巴道转移,血行转移较少。癌组织可直接蔓延或循淋巴管浸润而侵犯邻接组织。向下可侵犯阴道,向上可蔓延至宫体,向两侧可以延及宫旁及盆壁组织,可因肿瘤压迫输尿管而引起肾盂积水。晚期可侵犯膀胱和直肠。淋巴道转移是宫颈癌最重要和最多见的转移途径。一般是通过宫颈旁淋巴管先转移至闭孔、髂内、髂外等淋巴结,而后再转移至髂总、深腹股沟或骶前淋巴结(图13-5)。晚期患者可转移至锁骨上淋巴结。血行转移在宫颈癌很少见,其最多见的部位是肺、骨及肝。
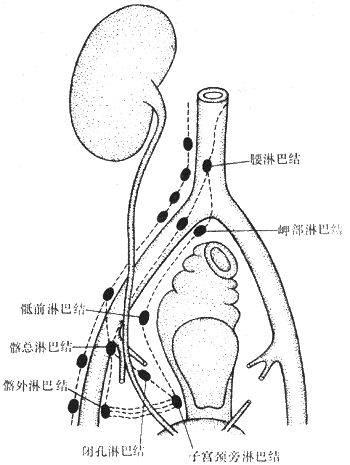
图13-5 子宫颈癌淋巴道转移示意图
第三节 子宫体疾病
一、子宫内膜增生症
子宫内膜增生症(endometrial hyperplasia)临床表现为功能性子宫出血,主要症状为月经不规则、经期延长和月经量过多。大部分病人发生于更年期或青春期。其发生与卵巢雌激素分泌过多而孕酮缺乏有关。
【病变】
肉眼观,一般可见子宫内膜普遍增厚,可达0.5~1cm,表面光滑,柔软,也可呈不规则形或息肉状。镜下,可分4种类型:①单纯型,子宫内膜腺体及间质均增生,腺体明显增多、大小不一、分布不均(图13-6)。偶见腺体扩大成囊,腺上皮细胞呈柱状,缺乏分泌,往往排列成假复层。核分裂像常见。间质细胞排列紧密。②囊腺型,以增生腺体呈明显囊性扩张为特征。典型病例肉眼可见在增厚的内膜中有散在小孔形成,因此称之为瑞士干酪样增生。镜下,内膜腺体形状多样,大小极不一致,小者如增生早期的腺体,大者直径可为小的数倍至数十倍,大小腺体皆衬以假复层高柱状或立方上皮,并缺乏分泌现象。间质细胞丰富,胞浆少,核浓染。③腺瘤样型,以腺体增生而密集排列和间质稀少为特征,腺体数量远比前两型为多,结构也更加复杂,腺上皮向腺腔内呈乳头状或向间质呈出芽样增生。间质稀少。腺上皮细胞为高柱状,假复层,核空泡状,核分裂像常见,但无明显异型性。④不典型增生,组织结构与腺瘤样增生相似,腺体拥挤并呈不规则形、分支状或出芽样增生,间质明显减少,同时出现腺上皮细胞的异型性,细胞核大,染色质粗,核仁明显,上皮复层,失去极性,常见核分裂像。子宫内膜不典型增生有时很难与高分化腺癌鉴别,主要鉴别点是前者不见间质浸润。有人认为它是子宫内膜腺癌的癌前变化。
图13-6 子宫内膜增生症
腺体明显增多、大小不一、分布不均,腺上皮多层,间质增生,排列紧密
【临床病理联系】
子宫内膜增生症的子宫不规则出血是由卵巢滤泡不排卵所致。由于卵巢持续分泌雌激素,一方面引起子宫内膜增生,另方面抑制垂体前叶卵泡刺激素的分泌,终致卵泡因失去卵泡刺激素的支持而发生退化,雌激素分泌因而急骤下降,增生的子宫内膜由于雌激素突然不足而发生坏死脱落,引起子宫出血。
有些功能性子宫出血的病人,偶尔在更年期前或更年期可见到分泌期内膜,有人认为系更年期前激素不平衡,垂体促性腺激素产生过度或黄体功能过盛所致。临床可有月经过多表现,称排卵性功能性子宫出血。
二、子宫内膜异位症
当子宫内膜组织出现于正常子宫内膜以外的部位时,称为子宫内膜异位症。一般称子宫内子宫内膜异位症为子宫腺肌病,子宫外子宫内膜异位症为子宫内膜异位症。
(一)予宫腺肌病
子宫腺肌病(adenomyosis)是指子宫肌壁内出现子宫内膜腺体及间质,在异位的腺体及间质周围有增生肥大的平滑肌纤维。多发生于育龄期妇女,主要临床表现为子宫增大,痛经及月经过多。病因未明,有人认为是子宫内膜直接向肌层扩展的结果。病人常有雌激素增高及伴有子宫内膜增生症。妊娠分娩及过度刮宫等创伤可能会增加子宫内膜碎片进入肌层的机会,故多见于经产妇。
【病变】
肉眼观,分弥漫型及局灶型两种。弥漫型表现为子宫均匀增大;局灶型者子宫呈不规则增大,多见于子宫后壁,呈球形增大。切面可见在增厚的子宫壁中散在大小不等的小腔,有些小腔含血性浆液或巧克力样液,有时可见棕色含铁血黄素沉着,偶尔有较大的囊腔形成。小腔隙周围可见平滑肌纤维呈旋涡状排列,与平滑肌瘤相似,故有腺肌瘤(adenomyoma)之称,但与周围肌层的分界不如平滑肌瘤明显。
镜下,子宫腺肌病的诊断标准是,距子宫内膜基底层以下至少一个低倍(10×10)视野(大约2mm)深处的子宫肌层中出现子宫内膜腺体及间质,呈岛状分布,其周围有肥大的平滑肌纤维。异位的腺体往往呈增生期改变。这种内膜对雌激素有反应,而一般对孕激素比较不敏感,可能与其来自子宫内膜基底层有关。少数腺体可呈分泌期改变。间质与正常子宫内膜间质相似。
(二)子宫内膜异位症
子宫内膜异位症(endometriosis)是指子宫内膜异位于子宫以外的组织及器官,如卵巢、子宫直肠窝、输卵管、子宫浆膜、子宫韧带、直肠、膀胱、子宫颈、阴道、外阴、腹股沟、腹部手术瘢痕、盆腔淋巴结等。一般好发于青年妇女和不孕症妇女。主要症状为痛经。
最多见的子宫内膜异位症为卵巢子宫内膜异位症,常见于卵巢表面,且多为双侧性。因引起局部纤维组织增生而常形成结节状肿物。异位的子宫内膜在卵巢激素作用下,可发生周期性变化,随月经反复出血,在局部形成充满“血液”的囊性肿物,即所谓子宫内膜异位囊肿。囊内含棕红色粘稠的变性血性液体,似巧克力糊、称巧克力囊肿。镜下,在其囊壁内往往可找到子宫内膜腺体及间质。由于经血不断产生和聚集,囊肿可继续增大,囊内压不断增高,引起临床症状。囊肿也可破裂,引起腹腔出血和附近组织粘连。其次为子宫直肠窝子宫内膜异位症,常引起严重粘连,并往往形成紫褐色结节状肿物。
组织发生主要有两种学说,一为种植及移植学说,认为在行经期,经血倒流至输卵管,进入盆腔,使子宫内膜碎片种植在盆腔及腹膜表面;或经淋巴道、血道带到淋巴结和远隔部位;或在子宫切开或切除手术时人为地将子宫内膜移植到腹壁切口所致。另一种是体腔上皮化生学说,认为异位的子宫内膜组织是由于器官本身局部的体腔上皮化生而来,这种可能性较小。
三、子宫肿瘤
(一)子宫平滑肌瘤
子宫平滑肌瘤(leiomyoma)是女性生殖器官中最常见的一种良性肿瘤,多见于30~50岁妇女,20岁以下罕见,绝经后肌瘤可逐渐萎缩。其发生可能与过度的雌激素刺激有关。临床上多数患者可无症状,若出现症状,则表现为月经过多及局部肿块等。
【病变】
肉眼观,肌瘤可以生长在子宫任何部位,常位于子宫壁内(肌层内肌瘤)、浆膜下(浆膜下肌瘤)或粘膜下(粘膜下肌瘤)。可单发或多发,常为多个,其数目多少不等,常见为数个、乃至十数个或数十个,称多发性平滑肌瘤。肌瘤的大小可极为悬殊,小的在显微镜下才可检见,大的如成人拳大或更大,甚至可充满整个腹腔。肌瘤多呈球形或融合成不规则形,质较硬,界限明显,但无明显包膜(图13-7)。切面上,瘤组织常呈灰白色,编织状或旋涡状,当肌瘤生长较快或供血不足时,可发生各种继发性改变,如玻璃样变、粘液变、囊性变、水肿及出血、坏死等。
图13-7 子宫平滑肌瘤
多个肌瘤结节,位于肌壁内、粘膜下及浆膜下,境界分明,宫腔受挤压呈裂隙状
镜下,瘤细胞与正常子宫平滑肌细胞相似,但肌瘤细胞核比较密集,常排列成纵横交错的不规则束状或成编织状。核大多呈长杆状、两端钝圆或圆锥形,染色质纤细。肌细胞间有不等量的结缔组织。每10个高倍(400倍)视野核分裂像少于5个者一般为良性。有少数病例瘤细胞核增多、致密,核大活跃,染色质粗,无核分裂像,称细胞性平滑肌瘤(cellular leiomyoma)。子宫平滑肌瘤的恶变率很低,据报道为0.2%~0.5%,多见于年龄较大、生长较快与较大的肌瘤。如果核分裂像每个高倍视野达10个以上或有肌层及血管浸润者应诊断平滑肌肉瘤。
(二)子宫体癌
子宫体癌又称子宫内膜癌(carcinoma of endometrium),较常见,占女性生殖道恶性肿瘤的20%~30%。近年来子宫体癌的发病率有上升趋势。多发生在50岁以上绝经期和绝经期后妇女。病因未明,一般认为与雌激素长期持续作用有关。主要临床表现为不规则阴道流血。
【病变】
肉眼观,分弥漫型及局限型两种。弥漫型的子宫内膜呈弥漫增厚,不规则形、息肉或菜花状,质脆,常见出血、坏死或溃疡形成。癌组织浸润肌层,深浅不一(图13-8)。局限型多位于宫底或宫角,后壁多于前壁,常呈息肉状伴肌层浸润。
图13-8 子宫体腺癌(弥漫型)
癌组织弥漫浸润子宫壁,且部分突入子宫腔中形成大不不等的乳头状肿块
镜下,子宫内膜癌多数为分化较好的腺癌。腺体数目增多,形状和大小不一,排列紊乱,腺体呈“背靠背”形式。腺体由单层或复层细胞组成,形成条索状“搭桥”。间质极少。有时腺上皮增生形成实体性癌巢。癌细胞呈柱状或多角形,可有不同程度的分化。胞浆中等量,淡染;核大小不一,染色质增多而深染,核仁明显,可有正常或病理核分裂像。根据分化程度,子宫内膜癌可分为3级:Ⅰ级(高分化腺癌),腺体较规则,同单层细胞组成,细胞排列紧密有的成假复层或复层。核分裂像少见(图13-9)。Ⅱ型(中分化腺癌),腺体不规则,有较多腺样结构或小腺体形成,可有少量实性区。癌细胞分化较差,核分裂像易见。Ⅲ级(低分化腺癌),腺体结构极少见,癌巢多呈实性片块状,细胞异型性明显,核分裂像多见。有些腺癌组织中可见良性化生的鳞状上皮团,称腺棘癌(adenoacanthoma)。如果腺癌内混有恶性鳞状上皮,称腺鳞癌(adenosquamous carcinoma),多见于分化较差的腺癌。少数子宫内膜癌含有透明细胞,或含粘液的细胞。有些可形成乳头状子宫内膜癌。
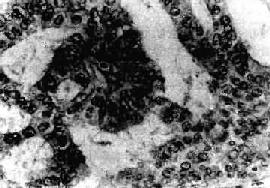
图13-9 子宫内膜腺癌
呈腺样结构,癌细胞呈柱状和立方形,核异型明显,排列紊乱
【扩展和转移】
一般,子宫内膜癌生长较缓慢,局限在子宫内膜的时间较长,但也有极少数发展较快者。其转移途径主要是淋巴道转移、直接蔓延,晚期也可有血行转移。
1.淋巴道转移 宫底部的癌多转移至腹主动脉旁淋巴结;癌在子宫角时可沿圆韧带的淋巴管至腹股沟淋巴结;子宫下段及扩散到宫颈管的癌灶,与宫颈癌的淋巴道转移途径相同,可至子宫旁、髂内外和髂总淋巴结。
2.直接蔓延,向上经子宫角至输卵管;向下至颈管、阴道;向外经肌层浸润至浆膜面而蔓延至输卵管、卵巢,并可广泛种植在腹膜、子宫直肠窝有大网膜等处。
3.血行转移,晚期患者可经血行转移至肺、肝有骨等处。
第四节 滋养层细胞肿瘤
滋养层细胞肿瘤包括葡萄胎、侵蚀性葡萄胎和绒毛膜癌。
一、葡萄胎
葡萄胎亦称水泡状胎块(hydatidiform mole),其病因及本质尚未完全阐明。目前多数学者认为葡萄胎是一种良性滋养层细胞肿瘤,但仍有少数学者认为葡萄胎是一种病理性妊娠,可能是胚胎缺陷或胚胎早期死亡后绒毛产生继发性退变的结果。
葡萄肿的发病率有明显的地区差异,欧美国家比较少见,约为1:2000妊次,而东南亚国家的发病率约10倍于欧美国家。我国1979年全国普查发病率为290/10万。妊娠次数与葡萄胎数之比为1290:1。上海地区调查发病率为1910:1。
【病变】
肉眼观,典型的葡萄胎形状极似葡萄。由于大部或全部胎盘绒毛间质水肿而显著肿胀,形成薄壁透明囊性葡萄样物,内含清液。大小不一,直径0.5~2、3cm,它们之间有细蒂相连,形如葡萄串(图13-10)。多数病例(约70%)所有绒毛都形成葡萄状,没有胎儿或其附属物,称完全性葡萄胎(complete mole);较少数病例(约30%)部分绒毛形成葡萄状,仍有部分正常绒毛,且常伴有或不伴有胎儿或其附属物,称部分性葡萄胎(partial mole)。
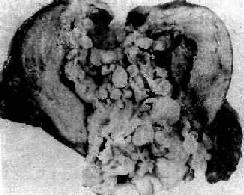
图13-10 葡萄胎
子宫体积增大,子宫腔中充满肿胀成葡萄状的绒毛
镜下,葡萄胎镜检有3个特点:绒毛因间质水肿而增大,并有水泡形成;间质血管消失或稀少;滋养层细胞有不同程度的增生。在这些特点中以滋养层细胞增生最重要。增生的滋养层细胞可为合体细胞或细胞滋养层细胞,大多两者混合并存,并具有一定的异型性(图13-11)。完全性葡萄胎往往增生明显。部分性葡萄胎常为局限性、轻度增生。近年来,有不少学者企图根据滋养层细胞的增生及分化程度来分级,以预测葡萄胎患者的预后,有的作者认为根据滋养层细胞增生及分化程度可以预测恶变的机会,但也有人持否定态度。
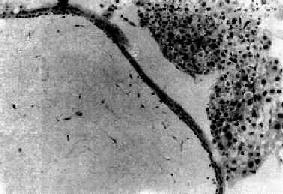
图13-11 葡萄胎
绒毛肿大,间质水肿,血管消失,滋养层上皮增生(图右侧)
【临床病理联系】
由于胎盘绒毛肿胀,子宫明显增大,致超出正常妊娠月的子宫大小。胚胎常早期死亡,故子宫虽可大如5个月妊娠,但听不到胎心音。由于滋养层细胞显著增生,胎盘激素分泌显著增多,其中以绒毛膜促性腺激素增多意义最大,能反映肿瘤的生长状态。葡萄胎一经确诊后应立即予以清除,大多数患者经彻底清宫后即可痊愈。约15%可恶变为侵蚀性葡萄胎,3%恶变为绒毛膜癌。部分性葡萄胎的恶变率很低。
二、侵蚀性葡萄胎
侵蚀性葡萄胎(invasive mole)也称恶性葡萄胎(malignant mole),多数继发于葡萄胎之后。侵蚀性葡萄胎与良性葡萄胎不同之处是前者水泡状绒毛侵入子宫肌层,且往往侵入肌层深层,引起组织破坏,甚至穿破肌壁引起大出血,并可转移至邻近或远处器官。此外,滋养层细胞增生及异型程度亦往往较良性葡萄胎显着。
临床表现主要为在葡萄胎排出后,血或尿妊娠试验持续不正常;阴道持续或间继不规则流血;胸片示肺内往往有转移灶;有时阴道可出现紫蓝色结节,破溃时可发生反复大出血。近年来由于化学疗法的进展,治疗侵蚀性葡萄胎有很好的疗效。
三、绒毛膜癌
绒毛膜癌(choriocarcinoma)简称绒癌,是滋养层细胞的高度恶性肿瘤。约50%继发于葡萄胎,25%继发于自然流产,20%以上发生于正常妊娠,5%以下发生于早产或异位妊娠等。主要临床表现是在葡萄胎、流产或足月产后阴道持续不规则流血,血及尿中HCG浓度显着升高。
【病变】
肉眼观,子宫不规则增大,柔软,表面可见一个或多个紫蓝色结节。切面可见肿瘤呈暗红色、出血坏死的肿块充塞宫腔(图13-12),或为多数结节浸润子宫肌层,常达浆膜,使子宫体积显着增大,或呈弥漫息肉状布满子宫内膜面,或在内膜和肌层内有小出血灶。肿瘤的特点是呈暗红色、质脆而软的出血、坏死病灶。
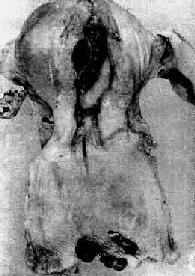
图13-12 子宫绒毛膜上皮癌及阴道壁转移
原发癌位于子宫底部,呈暗红色(图中为黑色)血肿样结节,并转移到阴道壁
镜下,成片增生及分化不良的滋养层细胞侵入肌层和血管。瘤组织由分化不良的两种滋养层细胞组成,即细胞滋养细胞和合体滋养细胞。细胞滋养细胞胞浆丰富、淡染,细胞境界清楚,核空泡状。合体滋养细胞体积大、胞浆红染并互相融合,核椭圆形。这两种细胞排列紊乱(图13-13)。不同肿瘤中这两种细胞所占比例不同,有的以细胞滋养细胞为主,有的以合体滋养细胞为主。核分裂像常见。绒癌组织无间质,常呈广泛出血坏死,不形成绒毛结构。如发现有绒毛,即使是退化的绒毛,也应诊断为侵蚀性葡萄胎。
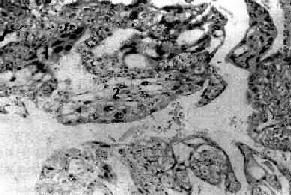
图13-13 绒毛膜癌
癌组织没有间质,癌巢由于两种细胞组成:1.合体细胞样细胞;2.细胞滋养层细胞样细胞
绒癌易侵入血管,故主要为血行转移,最多见转移至肺,其次为阴道、脑、肝、脾、肾、肠等。自应用化疗后,绒癌的死亡率已显著下降。
第五节 输卵管疾病
一、输卵管炎症
输卵管炎症比较常见,有急性、慢性和结核性输卵管炎等。
急性输卵管炎(acute salpingitis)多发生于分娩后、流产后或刮宫术后感染。常由化脓菌感染引起。病变常为双侧性。输卵管充血、肿胀、浆膜面可见纤维素性及脓性渗出物。及时治疗可完全治愈。若病变迁延,可转为慢性输卵管炎(chronic salpingitis),伞端可因炎症而闭锁,继之管腔扩张积脓,称输卵管积脓(pyosalpinx)。如输卵管卵巢粘连一起可形成输卵管卵巢脓肿。脓液吸收后遗留清液,可形成输卵管积水(hydrosalpinx)或输卵管卵巢囊肿。
结核性输卵管炎(tuberculous salpingitis)常继发于身体其他部位的结核,并经常成为女性生殖器官结核的原发部位。结核菌从输卵管以下蔓延到子宫体和子宫颈,引起子宫内膜结核和子宫颈结核。少数可引起阴道和卵巢结核。
二、输卵管妊娠
受精卵在子宫内膜以外的部位植入和发育生长,称异位妊娠(ectopic pregnancy),可发生于输卵管、卵巢、子宫颈、腹腔等处,最常见的是输卵管妊娠(tubal pregnancy)。导致输卵管妊娠的原因有3方面:①由于输卵管本身的疾病,如慢性输卵管炎,使受精卵在输卵管内受阻,延缓了进入子宫的时间而导致受精卵在输卵管内着床。②输卵管由于子宫内膜异位或蜕膜反应,增强了接受受精卵的能力,使受精卵在卵管内着床。③受精卵本身的因素,如受精卵在经过输卵管的过程中还不成熟或过成熟。
【病变】
输卵管妊娠约80%发生于壶腹部,其他如峡部、漏斗部及间质部都比较少见。妊娠处输卵管肿大,壁增厚,腔内含胚胎组织(图13-14)。由于输卵管壁薄,内膜的蜕膜反应弱,不适合受精卵生长发育,故一般在妊娠早期即发生流产,导致输卵管破裂出血,引起腹腔内出血,胚胎可随血液进入腹腔而死亡。极少数情况下,由于胎盘剥离不多,胚胎仍可在腹腔内继续存活,形成继发性腹腔妊娠。镜下,可见妊娠部位的输卵管粘膜固有膜呈蜕膜变化,并可见滋养层细胞及胎盘绒毛。如胚胎在妊娠较晚期死亡,胚胎可发生木乃伊化或钙化形成石胎(lithopedion)。

图13-14 输卵管妊娠
输卵管极度膨大,内含血块、胎盘组织和一小胎儿
第六节 卵巢肿瘤
卵巢肿瘤按其组织发生可分为三大类:①上皮性肿瘤,如浆液性肿瘤、粘液性肿瘤、子宫内膜样瘤及纤维上皮瘤等,这类肿瘤的性质可分为良性、交界性及恶性;②性腺间质肿瘤,如颗粒细胞瘤、泡膜细胞瘤及纤维瘤等;③生殖细胞肿瘤,如畸胎瘤、无性细胞瘤、内胚窦瘤及胚胎性癌等。
一、卵巢上皮性肿瘤
卵巢上皮性肿瘤是最常见的卵巢肿瘤,来源于卵巢表面的上皮(体腔上皮),最常见的是囊腺瘤,主要包括浆液性和粘液性两种。
(一)浆液性肿瘤
1.良性浆液性囊腺瘤(serous cystadenoma)是浆液性肿瘤中最常见的一种,约占浆液性肿瘤的60%,多发生于20~40岁妇女,以单侧居多,也可双侧发生(约占20%)。
【病变】
肉眼观,多为圆形或卵圆形囊肿,囊内充满稀薄、清亮的浆液,体积大小不一,小者直径仅数厘米,大者可达儿头大或更大,表面光滑,多为单房性,少数可为多房性。囊内壁光滑为单纯性浆液性囊腺瘤;部分伴有乳头状突起,称为乳头状浆液性囊腺瘤(图13-15)。镜下,囊壁和乳头间质均由含血管的纤维结缔组织构成,被覆上皮呈单层低立方状、柱状、纤毛柱状或钉状,核多位于中央,染色质纤细,核仁缺如或不明显,无病理性核分裂像(图13-16)。有时在囊壁和乳头间质内可见圆形钙化小体(沙粒体)。
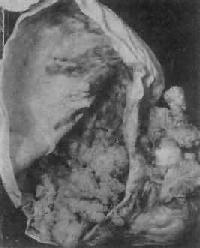
图13-15 卵巢乳头状浆液性囊腺瘤
肿瘤为单房性,囊壁内面及外表面(右侧)有乳头状肿块形成

图13-16 卵巢乳头状浆液性囊腺瘤
被覆囊壁的瘤细胞呈立方形,且呈乳头状生长,突向囊腔
2.交界性浆液性囊腺瘤(borderline serous cystadenoma)约占浆液性肿瘤的10%,其形态结构介于良、恶性浆液性囊腺瘤之间,属低度恶性,预后比浸润癌为好。
【病变】
肉眼观,与良性浆液性乳头状囊腺瘤相似,但乳头状突起往往比良性者丰富而广泛,常布满整个囊内表面,双侧发生率较高。镜下,主要表现为乳头上皮呈2~3层,乳头分支较稠密或有微乳头状突起,核异型和核分裂像易见(每高倍视野不超过1~2个),无间质浸润。
3.浆液性囊腺癌(serous cystadenocarcinoma),约占浆液性肿瘤的30%,为卵巢恶性肿瘤中最常见的类型,约半数为双侧性。患者以40~60岁妇女为最多。
【病理变化】
肉眼观,多数为多囊性,部分或大部囊内或囊外有乳头状突起,囊内多含混浊液体,乳头状物多为实性菜花状,常侵犯包膜并有出血坏死。镜下,乳头分支多或呈实心团块,上皮细胞增生多呈3层以上,细胞有明显异型性,核分裂像常见,包膜和间质均有浸润,沙粒体较多见。根据乳头状结构的分化程度可将其分为高分化、中分化和低分化3型:①高分化型(图13-17),多数乳头覆以不典型上皮,呈假复层,有一定的纤细间质;②中分化型,乳头结构仍可见,上皮细胞分化不良,呈多层,核分裂像增多;③低分化型,乳头很少,瘤细胞呈实心片块或条索,偶尔形成腺样结构,瘤细胞有明显异型性,包膜和间质浸润明显。
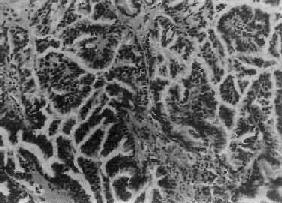
图13-17 卵巢乳头状浆液性囊腺癌
呈细乳头状结构,癌细胞多层,异型明显,乳头间质稀少
【转移】
有外生乳头的良性及交界性肿瘤都可以有盆腔或腹腔腹膜的种植。交界性瘤的种植转移更多见。多数浆液性囊腺癌在就诊时已有转移,转移部位为腹腔、盆腔浆膜层,一部分病例可发生淋巴结转移,包括盆腔、肠系膜淋巴结及锁骨上淋巴结等,极少数有远处转移,如肝、肺等。
(二)粘液性肿瘤
1.粘液性囊腺瘤(mucinous cystadenoma)是上皮性肿瘤中较常见的一种肿瘤。主要来源于卵巢表面上皮,向宫颈内膜上皮分化;另一来源是良性囊性畸胎瘤的单胚叶生长,其上皮和肠上皮相似,并可见杯状细胞。多发生于30~50岁妇女,多数为单侧,很少为双侧。
【病理变化】
肉眼观,囊性肿块大小不一,一般直径15~30cm,甚至50cm以上,小者直径仅1cm。圆或卵圆形,表面光滑,常为多房性,内含浓稠粘液。囊内壁光滑,很少有乳头。镜下上皮为单层高柱状粘液上皮,胞浆含清亮粘液,核位于基底部(图13-18),大小形状比较一致,染色质纤细,无明显核仁,亦无核分裂像。间质为纤维结缔组织。
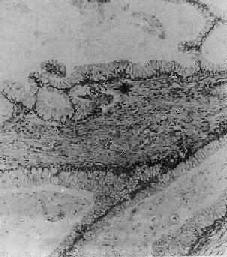
图13-18 卵巢粘液性囊腺瘤
瘤组织腺体扩大成囊,被覆囊壁的瘤细胞呈高柱状,核位于基底部,胞浆含大量粘液,(图中呈浅色透亮状)
2.交界性粘液性囊腺瘤(borderline mucinous cystadenoma)亦为低度恶性癌,形态结构介于良、恶性粘液性囊腺瘤之间。五年存活率为95%~98%。
【病理变化】
肉眼观与良性粘液性囊腺瘤无明显区别,但半数病例囊内壁可见乳头和包膜增厚,乳头或为简单分支,但多为生长活跃有复杂纤细分支的乳头。镜下,上皮高柱状,增生成2~3层,并失去极向,有轻或中度异型性,核分裂像可见。间质少,但无间质浸润。
良性及交界性粘液性囊腺瘤偶尔可自行穿破,使粘液性上皮种植在腹膜上继续生长并分泌粘液,形成腹膜假粘液瘤(pseudomyxoma peritonei)。
3.粘液性囊腺癌(mucinous cystadenocarcinoma),大部分患者年龄在40~60岁。
【病变】
肉眼观,肿瘤体积常较大,囊性或囊实性,表面光滑,常与周围器官粘连。20%为双侧性。多为多房性伴有实性区域,实性区为灰白色或质松脆的乳头状物,常伴出血坏死。囊内含有粘血性混浊液体。镜下,腺体密集,形状不规则,腺体上皮多超过3层,上皮细胞明显异型性,核仁明显,病理核分裂像易见。间质较少,可见包膜及间质浸润。根据上皮的分化程度可分为高分化、中分化和低分化3型:①高分化型,多数呈腺样结构,上皮高柱状,排列成3~4层以上(图13-19),含较多粘液,常有小乳头,可见一些核分裂像;②中分化型,腺体不规则,间质少,上皮异型性,排列乱,多层,核分裂像增多;③低分化型,腺样结构大部消失,上皮细胞分化差,核异型性明显,有多数核分裂像,常发生出血坏死,偶见粘液上皮。
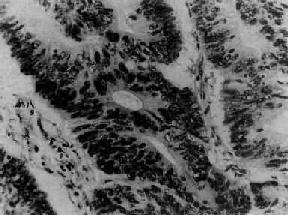
图13-19 卵巢粘液性囊腺癌
呈腺样结构,癌细胞高柱状,多层,异型性明显,核分裂像易见,仅少量粘液空泡形成
【扩散与转移】
卵巢粘液性囊腺癌可直接蔓延至阔韧带、输卵管和子宫,包膜浸润的癌细胞也可向腹腔内脱落或沿淋巴管扩散而转移,转移部位以盆腔、腹腔腹膜及各器官浆膜层为主,还包括大网膜、阑尾及对侧卵巢等。粘液性囊腺癌的5年存活率为46%~70%。
二、卵巢性腺间质肿瘤
本组肿瘤系来源于原始性腺中的性索及间质组织,包括粒层细胞瘤、泡膜细胞瘤、纤维瘤、支持细胞——间质细胞瘤、两性母细胞瘤及伴有环状小管的性索瘤等,大多数能产生性激素,引起相应的临床表现。最常见的是粒层细胞瘤和泡膜细胞瘤。
(一)粒层细胞瘤
粒层细胞瘤(granulosa cell tumor)约占卵巢肿瘤的1%~9%,好发年龄为45~55岁,少数也可发生于青春期前或幼女。肿瘤常产生雌激素,在青春期前表现性早熟、乳房增大、出现阴毛和初潮提前等;在青春期后则由于引起子宫内膜增生症而出现月经紊乱。
【病理变化】
肉眼观,肿瘤呈圆形、卵圆形或分叶状,大小不等,多数小于15cm,表面光滑,常有完整的包膜。切面多为实性,质地稍硬,肿瘤呈黄白色,有些可呈囊性变,常有坏死出血区。多为单侧性。镜下,瘤细胞多呈小圆形或多角形,胞浆少。核圆、椭圆或梭形,核膜清楚,核内常可见纵沟,核染色质细,可见1~2个核仁,核分裂像较少见。组织学类型有以下几种:分化较好的瘤细胞常排列成小卵泡型(图13-20),呈菊形团或小腺泡状,中央为粉染蛋白液或退化的细胞核,称Call-Exner小体;有的排列呈大卵泡型、梁柱型或岛状等;分化较差的瘤细胞常排列呈弥漫型或波纹状、脑回状或肉瘤样构型。
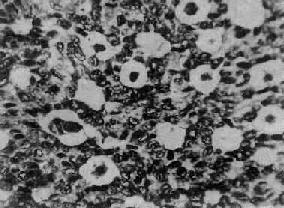
图13-20 粒层细胞瘤
瘤细胞排列成小卵泡型,部分细胞核呈现核沟
粒层细胞瘤为低度恶性肿瘤,预后尚好,晚期复发是该瘤的特点。5年存活率为80%~90%,10年存活率为70%左右。转移和复发多发生在腹腔内,远处转移很少。若浸润包膜或破裂,肿瘤可侵及邻近器官或对侧卵巢。
(二)泡膜细胞瘤
卵巢泡膜细胞瘤(theca cell tumor)比粒层细胞瘤少见,其发病率仅为粒层细胞瘤的1/3,占所有卵巢肿瘤的0.5%~1%。也有分泌雌激素的功能,常和粒层细胞瘤混合存在。发病年龄较晚,多数为绝经后妇女,发生在青春期前很少,不到1%。
【病变】
肉眼观,绝大多数为单侧,中等大小,与残留的卵巢组织界限清楚,但无包膜,质硬。切面实性,可有小囊,也有多房者。瘤组织因含脂质而呈浅黄色,间有灰白色纤维组织或类似纤维瘤的旋涡状结构。镜下,瘤细胞形态与正常泡膜细胞相似,胞体形态肥胖呈梭形,胞浆丰富淡染(图13-21)。细胞核卵圆形,无核分裂像。瘤细胞排列紧密,形成宽阔的束带状,呈不规则交错分布或漩涡状排列,其间被大小不等的间质结缔组织条索所分隔。用特殊染色法可见泡膜细胞内含有丰富的类脂质物质,可与纤维瘤鉴别;用银染色法可见网状纤维围绕每个细胞,此与粒层细胞瘤时出现于整个细胞团周围者不同。
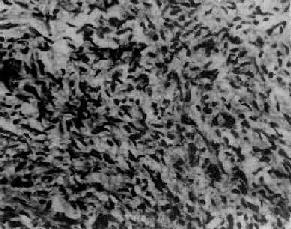
图13-21 卵巢泡膜细胞瘤
在卵巢泡膜细胞样瘤细胞中混杂一些空泡状黄体化细胞
泡膜细胞瘤基本为良性肿瘤,仅有个别复发者,但由于泡漠细胞瘤比粒层细胞瘤分泌更多的雌激素,所以并发子宫内膜癌的机会比粒层细胞瘤高,因此,术前诊断性刮宫以排除宫内膜病变是必要的。
三、卵巢生殖细胞肿瘤
卵巢生殖细胞瘤占卵巢肿瘤的15%~20%,20岁以下的卵巢瘤患者约60%属此类,所幸的是95%的生殖细胞瘤为良性囊性畸胎瘤。患者越年轻,所发生肿瘤的恶性程度可能越高。
(一)卵巢畸胎瘤
来源于卵巢多能的生殖细胞,其良恶性程度主要取决于瘤组织的成熟度,因此将畸胎瘤分为成熟型和未成熟型两大类。
1.成熟型畸胎瘤(teratoma,mature type) 多数为囊性,称囊性畸胎瘤,属良性,大多为单侧性。在卵巢生殖细胞瘤中最多见,占卵巢畸胎瘤的97%~99%,占所有卵巢肿瘤的20%。多发生于生育期妇女。
【病变】
肉眼观,肿瘤多为囊性、中等大小,表面光滑,囊内含毛发团及皮脂样物(图13-22)。囊壁较厚,内侧常有一处突起的结节或我称头节,表面被覆鳞状上皮,结节内常有毛发、牙或骨质等。肿瘤多数为单房。镜下,可见到三胚层各种类型的成熟组织,其中以皮肤、皮脂腺、汗腺、毛囊及脂肪最多见;其次为软骨、神经胶质、神经细胞、骨及呼吸上皮;其他如甲状腺、胃肠上皮及牙等较少见。
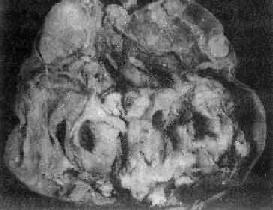
图13-22 卵巢囊性畸胎瘤
囊内充满含有毛发的黄色油脂样物
囊性畸胎瘤预后好,少数可发生恶性变,最常见者恶变为鳞癌。恶性变常发生在囊壁内头节附近,故检查时应注意取材部位。
2.未成熟型畸胎瘤(teratoma,immature type)此型较少见,仅占卵巢畸胎瘤的1%~3%。多见于25岁以下的年轻患者。
【病变】
肉眼观,肿瘤多为单侧性,体积一般较大,结节状,切面多为实性,夹杂有单个或多个大小不等的囊性部分。实性部分常为杂色,灰白、棕色或黄色,质软而脆,常有出血坏死。镜检可见由三胚层分化而来的未成熟和成熟组织混合组成。常见的未成熟组织为神经组织,如原始神经上皮和室管膜等结构及各种胚胎性组织,如胚胎性骨、软骨及肌肉等。皮肤组织较成熟型少见。其中可混杂一些各胚层的成熟组织。
未成熟型畸胎瘤常合并其他生殖细胞肿瘤,如内胚窦瘤、无性细胞瘤及绒癌等。一般说,肿瘤组织中未成熟组织及胚胎性组织的含量与临床恶性程度有关。病理上根据未成熟组织的含量,可分3级:①1级,主要为成熟组织,有少量未成熟组织,没有神经上皮或每一切片中神经上皮不超过一个高倍视野;②2级,有较多未成熟组织,每一切片中所含神经上皮不超过1~3个高倍视野;③3级,未成熟组织量多,每一切片中神经上皮超过4个高倍视野。
预后
未成熟畸胎瘤的预后与病理分级、临床分期密切相关。复发及转移者多为2、3级。转移的部位多在盆腔及腹腔内,远处转移极少。
(二)无性细胞瘤
无性细胞瘤(dysgerminoma),较少见,约占卵巢恶性肿瘤的1%~2%,多见于10~25岁年轻妇女。
【病变】
肉眼观,常为单侧性,约10%为双侧性。肿瘤多为圆或卵圆形,表面光滑,质韧。切面多为实性,有不同程度的出血坏死或囊性变,色灰红、暗红及棕黄色。镜下,与典型的睾丸精原细胞瘤非常相似,瘤细胞体积大而圆,比较一致,胞浆因含糖原而透明。核大,圆或卵圆形,大小形态一致,小泡状,核仁明显,核分裂像常见。瘤细胞排列成片块、巢状或条索。瘤细胞间常有不等量的淋巴细胞浸润及多少不等的纤维组织。由于肿瘤细胞形态和组织化学特性与未分性别的原始生殖细胞极相似,故命名为无性细胞瘤。
本瘤预后较好,对放射线敏感,5年生存率90%~95%。约5%病人对侧卵巢在显微镜下才能发现肿瘤,因此手术时必须进行活检,作冷冻切片检查。
肿瘤可直接蔓延扩散至盆腔附近器官及通过淋巴系播散到腹膜后及主动脉旁淋巴结等,晚期也可经血行播散到肺及肝等。
(三)内胚窦瘤
内胚窦瘤(endodermal sinus tumor)又称卵黄囊瘤(yolk sac tumor),来源于多能的生殖细胞,向胚外结构方向分化而形成的一种高度恶性的生殖细胞肿瘤。多见于儿童及年轻妇女,平均年龄18岁。除卵巢外,还可发生于骶尾、腹膜后、纵隔、松果体等处。
【病变】
肉眼观,肿瘤多为单侧性,以右侧为多,一般体积较大,平均直径为15~25cm,圆或卵圆形,表面光滑。切面多为实性,质较脆,灰白或粉白色,常有出血坏死及囊性变。镜检,组织形态较复杂及特殊,主要特征有下列几点:①网状结构,是最常见的形态,由星芒状的瘤细胞形成疏松网状结构(图13-23),其中有微型小囊或间隙,囊腔被覆扁平或立方细胞,这种结构类似胚外中胚层结构;②内胚窦样结构,立方或柱状的瘤细胞成单层排列,包绕毛细血管,形成一血管套样结构,这种结构横切面很像肾小球,称为Schiller-Duval小体,这种小体和大鼠胎盘内的Duval内胚窦结构相似,这种结构可能是由卵黄囊衍生演变而来;③多泡性卵黄囊样结构,特征是由扁平上皮、立方或低柱状的瘤细胞形成大小不等的囊腔,其间隔以致密的结缔组织或疏松的粘液样间质,这种结构与胚胎时期的卵黄囊(胚外内胚层)相似;④在瘤细胞内及瘤细胞间隙可见PAS阳性的大小不等的嗜酸性小滴,免疫组化显示这种小滴含有甲胎蛋白(AFP),正常情况下,卵黄囊可合成AFP;⑤可见腺样、乳头状及实体细胞团结构等。
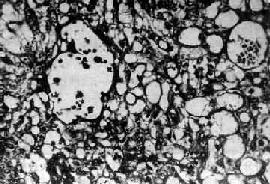
图13-23 内胚窦瘤
呈疏松网状粘液瘤样结构,其中有互相沟通、衬以上皮的腔隙及小囊腔,在瘤细胞内外可见嗜酸性玻璃样小球及基底膜样物
本瘤恶性度很高,预后差,对放射线不敏感。近年应用联合化疗后,3年存活率约13%。转移途径为直接浸润及种植扩散,偶有淋巴结转移,极少血行转移。
(四)胚胎性癌
胚胎性癌(embryonal carcinoma)为来自具有向胚外或胚内结构分化潜能的原始生殖细胞的一种未分化癌。较罕见,约占卵巢恶性生殖细胞瘤的5%。多见于儿童及青年,平均年龄为15岁。
【病变】
肉眼观,肿瘤一般为单侧性,体积较大,平均直径17cm,实性。切面棕褐或灰白色,常见出血坏死及囊性变。镜下,瘤细胞为大而幼稚的多形性细胞,胞浆中性呈不同程度的空泡状。核大而圆,空泡状,染色质粗,核仁明显,1至多个,核分裂像多见。瘤细胞排列呈实性巢状或腺泡、管状或乳头状,并常见散在的像合体滋养层细胞的多核巨细胞。免疫组化染色显示这种巨细胞含有绒毛膜促性腺激素(HCG),非巨细胞性瘤细胞内含有甲胎蛋白。
本癌为高度恶性,预后差,早期即可向局部器官和腹膜浸润,或通过淋巴道播散。5年存活率39%。
第七节 乳腺疾病
一、乳腺结构不良
乳腺结构不良(mammary dysplasia)又称乳腺纤维囊性病(fibrocystic disease of breast)或乳腺病(mastopathy)等,是妇女最常见的乳腺疾病,可发生于青春期后任何年龄,35~40岁为其发病年龄高峰。其病因一般认为是由于卵巢内分泌失调使性激素不平衡,主要是黄体酮减少而雌激素分泌过多,刺激乳腺组织过多增生所致。依其增生变化的形式,可分以下3种类型:
(一)乳腺组织增生
为本病早期病变,多见于30~40岁妇女,主要以乳房周期性疼痛为特征,伴有与月经周期有关的乳腺肿胀,单侧或双侧乳腺可触及弥散的颗粒状肿块。
【病变】
肉眼上一般无明显变化,乳腺组织切面有时可见散在小颗粒,偶见小囊。镜检可见乳腺小叶大小不一,形状不规则,末梢导管呈芽状增生,腺泡增多。管腔呈轻度扩张或有小囊腔形成,上皮细胞正常或增生为两层以上,有的可出现分泌现象。小叶间质纤维组织增生。这些病变通常在1~3年内可自行消失,部分病例可复发或发展为腺病。
(二)乳腺腺病
乳腺腺病(adenosis)以小叶腺泡、末梢导管和结缔组织增生为特征,小叶结构基本保存。依其不同发展阶段的不同组织学变化,大致可分为3型:①小叶增生型,主要表现为小叶的数目及小叶内腺泡数目略增多,因而小叶增大。上皮细胞没有明显改变或可呈双层、多层,小叶内导管可稍扩张。小叶内及其周围有多少不等的淋巴细胞浸润。小叶间结缔组织不增多或轻度增多。②纤维腺病型,一般由小叶增生型发展而来。主要特点是小叶内除末梢导管和腺泡增生外,间质结缔组织也有较明显增生(图13-24)。早期由于腺泡继续增生而使小叶增大;后期由于小叶内结缔组织明显增多,致使腺泡分散、变形,甚至萎缩。有的区域增生的肌上皮细胞没有基底膜,而是直接与胶原纤维接触,这可能说明肌上皮细胞有产生胶原纤维的可能,这时称之为硬化性腺病(sclerosing adenosis)。硬化性腺病有时与乳腺硬癌不易鉴别,但前者可见小叶轮廓、细胞无异型性、核分裂像罕见,是与乳腺癌的主要鉴别点。③小叶纤维化型,是腺病的晚期表现,一般为纤维腺病继续发展的结果。主要特点为小叶内间质纤维化和腺泡萎缩。小叶的轮廓有时存在,但也可消失,仅残存一些萎缩的导管。有时也可看到末梢导管扩张。
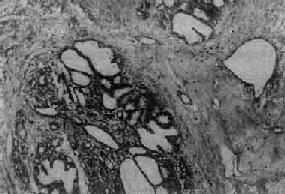
图13-24 纤维腺病
末梢导管、腺泡和间质均呈明显增生,部分腺泡及导管扩张,间质内有一些淋巴细胞浸润
(三)囊肿病
乳腺囊肿病(cystic disease)以小叶末梢导管和腺泡高度扩张成囊为特征。囊腔大小不等,多少不一,往往在肉眼上即可见到多人散在分布的小囊腔,直径在500~700μm以上者,称为囊肿病。镜检,囊内衬立方或柱状上皮及肌上皮细胞,在早期尚可见两层细胞结构,以后,部分或全部呈萎缩状态;有时在上皮萎缩消失处可见溃疡或肉芽组织形成;有的囊肿上皮则可增生,上皮细胞呈柱状,体积增大,有时增生的上皮构成筛状;或呈多发性乳头状瘤样增生,当若干扩张的导管和囊肿内均有乳头状增生时,则称为乳头状瘤病。此外,往往可见上皮呈大汗腺化生,细胞体积大、胞浆丰富、嗜酸性,PAS反应阳性。这种大汗腺化生往往是良性的标志,不要误诊为癌。
现在一般认为腺病与癌的关系不大,囊肿病伴有增生性病变时,癌变的机会较多,属于癌前病变。
二、乳腺癌
乳腺癌很常见,我国乳腺癌的发病率仅次于子宫颈癌而居女性癌瘤的第二位,近年来有不断增加的趋势。常发生于50岁左右的妇女,20岁以前很少见。患者女性亲属中乳腺癌的发病率高于常人2~3倍。其发生原因尚未完全阐明,一般认为可能与雌激素长期作用有关。本癌半数以上发生于乳腺外上象限,其次为乳腺中央区和区上象限。
【类型】
乳腺癌形态结构十分复杂,类型很多。一般根据组织发生和形态结构而将其分为导管癌、小叶癌和特异性癌三大类型:①导管癌,较多见,来源于乳腺导管系统,特别是末梢导管,包括非浸润性导管内癌及浸润性导管癌;②小叶癌,较少见,又称腺泡内癌,来源尚未完全确定,有人认为系起源于肌上皮细胞,也有人认为发生于小叶内导管,包括非浸润性的小叶原位癌及浸润性小叶癌;③特殊型癌,少见,为具有特殊形态结构的一类乳腺癌,如粘液癌、大汗腺样癌、腺样囊性癌、鳞状细胞癌及炎性癌等。
(一)导管内癌
多发生于乳头下乳晕周围,近半数病人在局部可扪及大小不等的肿块或境界不清的肥厚组织。肿块与皮肤无粘连。部分病人表现乳头溢液。由于癌在导管内生长并充满导管腔,致使导管扩张,因而在切面上可见导管为条索状(纵切)或小结节状(横切),灰白色或黄灰色,挤压之可从导管中挤出蠕虫样的半固体性物质。镜检,癌细胞位于扩张的导管内,基底膜完好。癌细胞大小形状不一,细胞分化程度各例不等,分化较高者,核分裂像少见;分化较低的病例,细胞异型性明显,核分裂像多见。其组织结构多样,癌细胞可在扩张的导管内排列成实心团块、乳头状、筛状、小管状,部分病例在管内实体细胞团中央可发生大片坏死,称粉刺性管内癌。
(二)浸润性导管癌
由导管内癌发展而来,为乳腺癌中最常见的类型,约占乳腺癌的50%~80%,以40~60岁妇女为最多见。
【病变】
肉眼观,肿块一般较小,直径多为2~3cm,质硬,边缘不整,常可见到灰白色癌组织呈放射状侵入邻接纤维脂肪组织内。如果癌瘤位置浅,则可侵犯皮肤,与皮肤粘连并导致皮肤出现不规则浅表微小凹陷,呈橘皮样外观;如累及乳头,可出现乳头回缩、下陷现象(图13-25)。镜检,组织形态多样,癌细胞组成实体团块或腺样结构,两者常混合存在。其中多数病例主要由实体癌细胞团组成,称为实体癌。一般根据实体癌的癌实质与纤维组织间质比例的不同,又将其分为单纯癌(simple carcinoma),实质与间质量大致相等(图13-26);硬癌(scirrhous carcinoma),癌实质少而间质多;和不典型髓样癌(atypical medullary carcinoma),癌实质多而间质少,间质内常无淋巴细胞浸润(图13-27),癌细胞常呈多形性,核异型性明显,核分裂像易见。
图13-25 乳腺癌
切面癌组织灰白色,较大的肿块左侧形成卫星结节;乳头略下陷
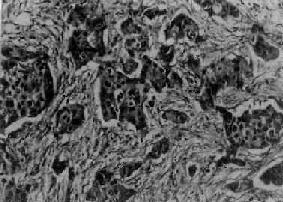
图13-26 浸润性导管癌(单纯癌)
癌细胞和间质量大致相等
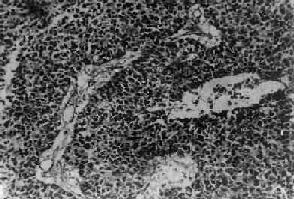
图13-27 乳腺髓样癌
癌细胞排列成片团状,间质少
(三)小叶原位癌
一般发生于绝经前妇女。临床上一般摸不到肿块,也无症状。标本肉眼观与一般小叶增生不易区别。镜检,癌变小叶体积增大,但小叶轮廓尚保存,小管高度扩张,其中充满单一松散排列的癌细胞。癌细胞呈圆形,大小形状较为一致,核圆形及卵圆形,核分裂像很少。基底膜完整。小叶原位癌经过一定时间可发展为浸润性小叶癌。
(四)浸润性小叶癌
由小叶原位癌突破小管或末梢导管基底膜向间质浸润所致。在大体上很像硬癌,肿块不规则盘状,与周围乳腺组织的边界不清。镜检的特征是癌细胞排列松散,呈条索状;有时为分散的单个癌细胞浸润于成束的纤维组织之间,这些细胞并无组成小叶的痕迹。癌细胞小或中等大小,多呈圆形、椭圆形或梭形,细胞的大小及染色较一致。有时可见从小叶原位癌向浸润性小叶癌过渡的形态。
(五)典型髓样癌
较少见。肿块体积常较大,直径4~6cm或更大,边界较清楚,质松软,灰白色,常杂以灰黄色或暗红色坏死出血区。镜下,癌实质多,间质少。癌细胞较大,圆形、卵圆形,胞浆嗜碱性,核大,染色质丰富,核仁不明显,分裂像较多。间质纤维组织稀少,其中常有淋巴细胞浸润。
本癌一般生长较慢,腋窝淋巴结的转移较少也较晚。预后比浸润性乳腺癌为佳,根治术后5年存活率近70%。
第十四章 男性生殖系统疾病
第一节 睾丸和附件炎症
一、肉芽肿性睾丸炎
肉芽肿性睾丸炎(granulomatous orchitis)比较少见,多发生于中年人。病因尚未阐明,患者常有睾丸损伤史,故可能为生殖细胞损伤后,产生或释放某种物质引起肉芽肿的形成,临床上可呈急性经过,睾丸呈明显的炎性肿痛,亦可进展缓慢,似睾丸肿瘤。
【病变】
肉眼观,睾丸体积增大,鞘膜呈局灶性或弥漫性增厚,鞘膜腔积液。切面可见病变睾丸呈弥漫或局限性灰白或黄褐色。镜下,细精管破坏,有由大量上皮样细胞、淋巴细胞、浆细胞、组织细胞和一些多核巨细胞与中性粒细胞形成的结核样结节,结节中央可见退化的精子,肉芽肿周围纤维组织增生,细精管的基底膜纤维性增厚,间质内有大量淋巴细胞和浆细胞浸润及纤维组织增生。
二、流行性腮腺炎性睾丸炎
流行性腮腺炎性睾丸炎(mumps orchitis)为约1/4的成人流行性腮腺炎可并发睾丸炎,而青春期前儿童患者合并睾丸炎者较少。病变的睾丸在急性期呈间质水肿及中性粒细胞、淋巴细胞和组织细胞浸润;细精管扩张,腔内含有同样炎细胞。40%~60%的病例可转变为慢性,细精管造精细胞消失,间质淋巴细胞浸润,并发生纤维化和玻璃样变,因此睾丸萎缩而丧失生精功能。所幸本病大多数为单侧而不影响生育能力,如双侧睾丸受累,则可引起不育。
三、睾丸树胶样肿
后天性梅毒第三期可侵犯睾丸,病变有两种,较常见的一种为树胶样肿(gumma);另一种为弥漫性炎症与纤维化。睾丸树胶样肿呈睾丸进行性肿大,无明显疼痛。直径一般为1~3cm,质较韧,切面为黄色不规则的坏死组织,周围包围一厚层纤维组织。镜下可见坏死组织中仍保持原有组织结构的痕迹,周围的纤维组织中有大量淋巴细胞和浆细胞浸润,亦可见多核巨细胞。树胶样肿旁和小血管内常可见闭塞性动脉内膜炎。弥漫性炎与纤维化的睾丸无明显肿大。镜检为弥漫性纤维组织增生,其中有大量淋巴细胞和浆细胞浸润,细精管萎缩。
四、精子性肉芽肿
精子性肉芽肿(spermatic granuloma)是由于附睾炎症或外伤,损伤输精管道致精子溢出至间质内而引起的病变。
【病变】
肉眼观,常在附睾的上极见一灰白色或灰黄色结节,直径一般在0.5~3cm。切面可见结节内含黄色或棕黄色物质。镜下,病变主要发生在附睾间质,早期主要为中性粒细胞和巨噬细胞浸润,中央为溢出或退化的精子;晚期病灶为结核样肉芽肿,由类上皮细胞、淋巴细胞和组织细胞组成,可见多核巨细胞,肉芽肿中央为退化的精子及细胞碎片,周围纤维母细胞增生,最后肉芽肿可能为纤维组织代替,形成玻璃样变的纤维性结节。
输精管精子性肉节肿可能与机械性阻塞(多有输精管结扎史)有关,常在输精管上形成多个硬结。其病变与附睾者相似。
五、附睾结核
结核常侵犯附睾,梅毒常侵犯睾丸。附睾结核常为单侧,多系由肺结核、肠结核、泌尿系结核及精囊前列腺结核等散布所致。
【病变】
肉眼观,附睾肿大。切面可见孤立或密集的灰黄色干酪样坏死灶。镜下可见典型的干酪样坏死及结核结节。晚期病变可发生纤维化及钙化。
第二节 睾丸肿瘤
睾丸肿瘤多为恶性,其发生率约占所有男性癌瘤的2%。根据组织发生可将其分为两大类,一类为起源于睾丸生殖细胞的肿瘤,包括精原细胞瘤、胚胎性癌、畸胎瘤、绒毛膜癌及内胚窦瘤等,较多见;另一类来自睾丸性索间质,包括睾丸间质细胞瘤和支持细胞瘤等,较少见。
一、精原细胞瘤
精原细胞瘤(seminoma)起源于睾丸原始生殖细胞,为睾丸最常见的肿瘤,多发生于中年以后,常为单侧性,右侧略多于左侧。发生于隐睾的机率较正常位睾丸高几十倍。本瘤为低度恶性。
肉眼观,睾丸肿大,有时可达正常体积的10倍,少数病例睾丸大小正常。肿瘤体积大小不一,小者仅数毫米,大者可达十余厘米,通常直径为3~5cm。由于睾丸白膜比较韧厚,未被肿瘤破坏,故通常睾丸的原来轮廓尚保存。切面瘤组织呈淡黄或灰黄色,实体性,均匀一致如鱼肉,其中往往可见到不规则坏死区。镜下,典型的精原细胞瘤有瘤细胞形态结构单一和间质内有淋巴细胞浸润两个特征,(图14-1)。瘤细胞弥漫分布或呈索状结构,细胞的形态一致,与正常精小管内精原细胞相似,瘤细胞大,圆形或多角形、境界清楚、胞浆透明,核大、位于中央,核膜及染色质较粗,有1~2个嗜酸性核仁,核分裂像不多见。间质为纤细的纤维组织或致密的胶原纤维,其中有多少不等的淋巴细胞浸润,有时可有淋巴滤泡形成。
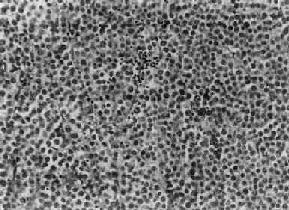
图14-1 精原细胞瘤
瘤细胞大、胞浆透明,核大,核仁明显,间质内有淋巴细胞浸润
少数精原细胞瘤的实质细胞大小不一,核粗大,分裂像较多,称为分化不良性或未分化性精原细胞瘤(anaplastic seminoma);另有少数精原细胞瘤的瘤细胞形态似不同成熟程度的精母细胞,称精母细胞型精原细胞瘤(spermatocytic seminoma),后者预后较好。
本瘤对放射治疗高度敏感。淋巴道转移较常见,血道转移较少发生。
二、胚胎性癌
胚胎性癌(embryonal carcinoma)起源于具有多分化潜能的原始生殖细胞,为高度恶性肿瘤,发病高峰在30~40岁,婴儿及儿童也可发生。
肉眼观,睾丸肿大,肿瘤常侵犯睾丸被膜及附睾。切面肿瘤实性,灰红或灰黄色,常有广泛出血坏死,偶见小囊腔形成。镜下,以癌组织结构的多样性为特征。瘤细胞为未分化的大小不一、形态不规则的细胞,细胞核大、染色深、染色质粗、核分裂像较多。细胞排列成各种不规则的条索状、网状、腺体状、圆柱状、乳头状,偶有囊腔形成。间质的形态很不一致,有的为胶原纤维,有的为肿瘤性原始间叶组织,有的为肉瘤样间质。
胚胎性癌常可与精原细胞瘤混合存在,或合并其他生殖细胞瘤。
胚胎性癌生长迅速,对放射线不敏感,预后较差,转移较早,多经淋巴道转移到髂内、髂总淋巴结。血道转移到肝、肺等处也较常见。
第三节 前列腺疾病
一、前列腺增生症
前列腺增生症(hyperplasia of prostate)又称前列腺肥大,多发生于50岁以上的老年人。其发病率依年龄增长而增加,70岁以上男性均有不同程度增生,但多数无症状。
【病因】
一般认为和体内雄激素及雌激素平衡失调有关。在正常情况下,雄激素主要促进前列腺上皮细胞的分泌,雌激素则主要促进前列腺间质结缔组织、平滑肌纤维和部分腺体增生。尿道周围部前列腺也称前列腺内区(包括尿道周围的中叶及部分侧叶,系由Müller管分化而来,此部分即所谓女性部),对雌激素敏感;而包膜下前列腺,也称前列腺外区(即所谓男性部)对雄激素敏感。在动物实验中给切除睾丸的大鼠注射雌激素,则前列腺增生;如同时注射雄激素或给未切除睾丸的动物注射雌激素均不形成前列腺增生。在人类,青春期阉割者不发生前列腺增生,所以雄激素的存在似乎是前列腺增生所必需的条件。人前列腺增生的原因可能和雄激素减少、雌激素相对增高的平衡失调有关。
【病变】
肉眼观,正常前列腺约栗子大,重约20g,增生的前列腺可达正常的2~4倍,甚至可达100g以上。切面见增生多发生于尿道两侧与后侧,或偶见只限于尿道后侧,将尿道压迫成一裂隙,并在膀胱的尿道开口处向膀胱内凸出(图14-2)。增大部的前列腺呈结节状,一般直径在0.5~2cm,灰白色,有纵横交错的条纹,其间夹杂有蜂窝状小孔,或小囊腔。切面的形态和增生的成分有关,如纤维、肌肉组织增生较显著时,则质地较实韧;如腺体增生较显著,则呈白色或灰黄色蜂窝状或囊性结构,用手指压迫时可有较多白色混浊的分泌物溢出于切面上。增生周围的前列腺组织被压迫而形成一假性包膜,所以能将增生的结节剥离出来。
图14-2 前列腺增生症
前列腺明显肿大,压迫膀胱颈部,且部分突入膀胱三角区;膀胱扩张肥厚,粘膜面可见代偿肥大的平滑肌条索呈梁状
镜下,可见前列腺的腺体、平滑肌和纤维结缔组织呈不同程度增生(图14-3)。一般认为前列腺增生是先在尿道两侧的粘膜下,纤维及平滑肌增生,形成多数的小结节状,以后腺体也相继增生,夹杂于增生的平滑肌与纤维组织之间而逐渐形成大小不等的、由纤维及肌组织包绕的腺体结节。增生的腺体腺泡数目增多,体积也呈不同程度扩大。上皮细胞呈柱状或立方形,核位于基底部,也可形成乳头状突入腺泡腔内。腺泡腔内有分泌物及脱落的上皮细胞,偶可见淀粉样小体。纤维及平滑肌细胞肥大、增生,包绕或穿插于增生的腺体之间,形成宽窄不一的间隔。间质中可见多少不等的淋巴细胞浸润。
前列腺增生常引起排尿障碍和继发感染,约有半数患者需要进行治疗才能解除痛苦。
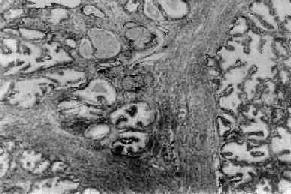
图14-3 前列腺增生
腺体平滑肌和纤维组织均呈明显增生,有些形成乳头状突入腺泡腔内,有些腔内含有分泌物
二、前列腺癌
前列腺癌(carcinoma of prostate)多发生于60岁以上的老年人,在我国远较欧美国家少见。美国报告前列腺癌的死亡率占恶性肿瘤死亡率的第二位,在我国前列腺癌的发生率只占恶性肿瘤的0.3%左右。
【病因】
前列腺癌的病因尚不十分清楚,一般认为激素特别是雄激素可能起重要作用。长期以来各家对前列腺癌与前列腺增生之间的关系看法不一。目前大多数学者认为两者间无明显关系,因为前列腺癌很少发生于良性增生的前列腺内。在组织发生上增生与癌的发生部位也不相同,前列腺增生多发生于尿道周围部的前列腺组织(即前列腺内区);而前列腺癌几乎都开始发生于前列腺的包膜下部(即前列腺外区),该部组织对雄激素敏感,高水平的雄激素可使该部增生。据统计,阉人不发生前列腺癌,这些都支持雄激素和前列腺癌有关。但前列腺癌患者多为老年人,睾丸产生的雄激素已经降低,所以仍有不支持此说的根据。有人假设前列腺癌发展很慢,可以长期处在潜伏状态,可能在雄激素尚处于较高水平时即已发生小的癌灶,发展到老年才出现症状,这一假设尚有待进一步验证。
【病变】
肉眼观,前列腺癌初期为单个或多数的硬结节,其前列腺可以增大,也可正常大小。早期病灶几乎都发生于包膜下,其中大多数发生于后叶,其次是两侧及前叶的包膜下,而发生于中叶者极为少见。晚期肿瘤可扩展到全部前列腺,使前列腺明显增大而质地变硬。切面灰白色夹杂以多少不等的纤维性条纹或间隔,也可呈均质性夹以不规则的黄色区域。
镜下,97%的前列腺癌均为腺癌,少数为移行细胞癌和鳞状细胞癌。依其分化程度可分为高分化、中分化和低分化3型。高分化前列腺癌最多见,癌细胞排列成大小不等的腺样结构,颇似前列腺增生腺体,但癌细胞体积较小,核较深染,上皮细胞往往呈多层排列并较不规则(图14-4),有时可呈乳头状腺癌或腺泡腺癌结构,并常可见癌组织向间质浸润生长;中分化腺癌全部或部分呈腺样结构,但腺体排列较紊乱,核异型性较明显,且有时形成筛状结构;低分化腺癌的癌细胞一般较小,排列成实体团块或条索,腺腔样结构很少(图14-5)。多数病例乃由上述多种组织结构混合组成。
图14-4 前列腺癌(高分化型)
腺体密集,癌细胞体积较小,核深染,上皮细胞呈多层排列并较不规则,可见间质浸润
图14-5 前列腺癌(低分化型)
癌细胞异型明显,并呈筛状结构
【转移及扩展】
前列腺癌的蔓延和转移与癌细胞分化程度有一定关系。高分化腺癌蔓延和转移较晚,可长期局限于前列腺内,预后较好。分化较差的腺癌可直接侵犯周围器官,如膀胱底、精囊腺、尿道等,但很少直接侵入直肠,因癌组织不易穿透直肠膀胱筋膜。前列腺癌的淋巴结转移比较常见,最常侵犯的淋巴结有髂内、髂外、腹主动脉旁、腹股沟等淋巴结,也可侵入胸导管、锁骨下淋巴结等处。血行转移可转移至骨、肺、肝等处,特别是腰椎、骨盆及肋骨的转移较常见。骨转移的途径有认为是经肺循环后再散布到全身的骨及肝组织,也有认为可经脊椎静脉丛直接转移至腰椎。
前列腺癌可分泌酸性磷酸酶,临床上常以此作为前列腺癌的一个检测指标。
第四节 阴茎癌
阴茎癌(carcinoma of penis)为常见的男性生殖系统的恶性肿瘤之一。多发生于中年人,平均年龄为30岁。较阴茎乳头状瘤的患者大10岁,所以它在初期可能为乳头状瘤,经若干年后转移为鳞状细胞癌。患阴茎癌者绝大多数皆有包皮过长。
阴茎癌多发生于包皮内面、阴茎头和冠状沟等处。早期病变可呈湿疹样、小乳头状、确实的红斑或白斑,尔后逐渐增大,局部隆起,表面呈菜花状或溃疡状,溃疡底高低不平,边缘不整且常隆起如围堤状,并常合并感染而有恶臭。切面上可见灰白色癌组织向下浸润生长,有时累及海绵体。晚期可直接蔓延到阴囊及会阴部。镜下,绝大多数阴茎癌为高分化鳞状细胞癌,有明显细胞间桥和角化珠形成。
阴茎癌转移发生较早,且大多沿淋巴道转移到腹股沟淋巴结,远处转移很少见。
阴茎鳞状细胞癌应与阴茎巨大尖锐湿疣鉴别。后者体积常较大,形成菜花状充满于包皮内,有时可穿出包皮或压迫阴茎头引起海绵体萎缩或破坏,可有继发感染,形成溃疡,而误认为阴茎鳞状细胞癌。但巨大尖锐湿疣的乳头隆起较大,棘细胞层明显增厚,角化不全显著,在上皮的中、表层可见空泡细胞,各层细胞的分化及极向保持良好,基底膜完整,其下间质内有较多淋巴细胞浸润等,可与阴茎癌鉴别。但阴茎巨大尖锐湿疣也有可能恶变为鳞状细胞癌。
第十五章 内分泌系统疾病
内分泌系统包括各内分泌腺体及弥散分布的神经内分泌细胞(即APUD细胞)。激素的合成与分泌一方面受神经系统的调控,同时也受下丘脑-垂体-靶器官之间的调节机制所控制。下丘脑的神经内分泌细胞分泌多种肽类激素,控制垂体许多激素的合成与分泌,垂体的激素又控制着靶器官激素的合成与分泌;反过来,靶器官所分泌的激素在血中的水平又对垂体及下丘脑相关激素的合成及分泌起反馈调节作用。通过上述调节,保持着各种激素的水平相对恒定。
各内分泌器官的肿瘤、炎症、血液循环障碍、遗传疾病及其他病变均能引起该器官激素分泌的增多或不足。但由于有上述调节机制,机体的激素水平仍然可以保持在正常范围内,只有超过了机体的调节能力,或者调节机制异常,机体内的激素水平才会失去平衡,临床表现为相应器官功能亢进或低下。可见内分泌系统疾病实际上包括内分泌器官的病变和由此引起的相应靶器官腺体的增生肥大或萎缩。例如垂体破坏性病变引起ACTH及TSH分泌不足,可导致肾上腺皮质及甲状腺腺体的萎缩及功能低下;垂体腺瘤时某种激素常分泌增多,引起靶器官腺细胞的肥大增生和激素分泌增多,后者又可反馈性抑制腺瘤外垂体正常部份某种激素的合成、分泌,并出现该腺细胞的萎缩;如果靶器官遭到破坏致功能低下,血中该激素水平下降,通过反馈机制使垂体相关激素分泌增多,进而使靶器官未受累的腺体肥大增生。
第一节 下丘脑和垂体疾病
下丘脑和垂体在解剖与功能上有密切关系。下丘脑与神经垂体实际为一个解剖、功能单位。下丘脑的视上核和室旁核神经细胞的轴突,经漏斗进入神经垂体的神经部(即垂体后叶),该神经细胞合成加压素(即抗利尿激素antidiuretic hormone,ADH)及催产素(oxytocin,OT),其分泌颗粒沿轴突运送到神经部,然后释放。下丘脑结节漏斗核等处的神经细胞,合成多种释放激素及抑制激素,其分泌颗粒在漏斗处释放入血,调节腺垂体功能。腺垂体包括远侧部(即垂体前叶)、中间部及结节部。垂体前叶在HE染色切片中可见三种细胞:嗜酸性细胞、嗜碱性细胞及嫌色细胞。电镜下,在细胞的胞浆中均可见有或多或少,不同大小的分泌颗粒,用免疫细胞化学方法可将前叶的细胞进一步分为:①促生长素细胞,分泌生长素(GH);②催乳素细胞,分泌催乳素(PRL)。以上两种细胞多嗜酸性;③促甲状腺素细胞,分泌促甲状腺素(TSH);④促性激素细胞,分泌促卵泡素(FSH)及促黄体素(LH);⑤促肾上腺皮质激素细胞,分泌促肾上腺皮质激素(ACTH)及促脂解激素(LPH)。第3、4、5种细胞多嗜碱性。大约有一半的嫌色细胞有稀少的分泌颗粒,另一半不见分泌颗粒,无内分泌功能。中间部及结节部也有一些含分泌颗粒的细胞,其功能尚不太清楚。
一、下丘脑及垂体后叶疾病
下丘脑-垂体后叶轴任何部位发生功能性或器质性病变时,不论其病因及病变性质如何,都会引起植物神经功能紊乱及内分泌功能异常,致使一种或数种激素的分泌过多或减少,临床上表现为各种综合征,例如尿崩症、性早熟症、肥胖性生殖无能综合征等。
(一)尿崩症
尿崩症(diabetes insipidus)由于ADH缺乏,使肾远曲小管对水分再吸收功能显着降低,因而表现为多尿(一昼夜可排尿5~20L)、尿比重显着低下(多在1.001~1.005)及由于大量水分丧失而出现的烦渴和多饮。其病因多为下丘脑-垂体后叶轴的肿瘤、外伤及感染引起的脑炎或脑膜炎;少数病因不明,找不出器质性病变。另外还有一种肾性尿崩症,是肾小管对ADH缺乏反应能力所致。
(二)性早熟症
性早熟症(precocious puberty)表现为女孩6~8岁、男孩8~10岁前出现性发育,病因为脑肿瘤、脑积水或遗传异常,使下丘脑过早分泌促性腺激素释放激素。
(三)肥胖性生殖无能综合征
肥胖性生殖无能综合征(dystrophia adiposogenitalis)表现为生殖器官、生殖腺体发育不全,无生殖能力,第二性征差,明显肥胖,常并发智能及精神上的缺陷。病因多为颅咽管瘤、垂体嫌色细胞瘤、神经胶质瘤及脑膜炎等引起的丘脑破坏,进而引起促性腺激素分泌障碍及肥胖。
二、垂体前叶功能亢进
垂体前叶病变引起的功能亢进绝大多数见于前叶良性肿瘤即腺瘤。该腺瘤一般过多地分泌某一种激素,出现该激素功能亢进症状,偶尔同时分泌二种激素,如GH及PRL。肿瘤周围的正常细胞因受压而萎缩,分泌激素减少,临床表现为功能低下。
(一)垂体性巨人症及肢端肥大症
垂体生长激素细胞腺瘤分泌过多的生长激素,使生长激素介质增多,促进DNA、RNA及蛋白的合成,在青春期以前即骨骺未闭合时,引起垂体性巨人症(pituitary gigantism);在青春期后骨骺已闭合时,则引起肢端肥大症(acromegaly)。垂体性巨人症表现为骨骼、肌肉、内脏器官及其他组织的过度生长,致使身材异常高大,内脏器官也按比例增大,但生殖器官如睾丸、卵巢等发育不全,女性病人常无月经,有的并发糖尿病。肢端肥大症发病呈隐匿性,就诊时病程常已有数年至10年之久,表现为头颅骨增厚,下颌骨、眶上嵴及颧骨弓增大突出,鼻、唇、舌由于软组织增生而增厚变大,皮肤粗糙增厚,呈现特有面容;四肢肢端骨、软骨及软组织增生使手足宽而粗厚,手指及足趾粗钝,内脏器官亦肥大。约有半数患者伴有其他内分泌功能障碍,如高胰岛素血症、糖耐量减低、或性功能减退。
(二)催乳素过高血症
催乳素过高血症(hyperprolactinemia)一部份是由于垂体催乳激素细胞瘤所致,另一部份是下丘脑的病变或雌激素、多巴胺能阻断剂等药物所引起,临床表现为溢乳-闭经综合征(galactorrhea-amenorrhea syndrome),女性患者有闭经、不育及溢乳,男性患者有性功能降低,少数亦可溢乳。
(三)垂体性Cushing综合征
垂体性Cushing综合征主要由ACTH细胞腺瘤所引起,少数由于下丘脑异常分泌过多的皮质激素释放因子(corticotropine releasing factor,CRF)所致。由于ACTH分泌过多,使两侧肾上腺皮质增生,分泌过多的糖皮质激素,详见第四节。
三、垂体前叶功能低下
垂体前叶功能低下多由于腺垂体严重破坏所致,少数由于下丘脑遭肿瘤破坏,使各种释放激素的分泌减少所引起。垂体前叶功能低下多数表现为全部激素的分泌低下,少数只表现为某一种激素的分泌低下。相对常见的综合征有:
(一)Sheehan综合征
Sheehan综合征是分娩后垂体坏死,使垂体前叶激素全部分泌减少的一种临床表现。多由于分娩时大出血或休克引起。妊娠期垂体增生肥大,压迫自身的血管,血流供应相对不足,因此对任何原因引起的局部血液灌注降低十分敏感,易发生梗死(图15-1)。当坏死面积超过垂体前叶的60%,可出现轻度功能障碍,超过90%时出现功能衰竭。典型病例在分娩后2~3周出现乳腺急骤退缩,乳汁分泌停止,以后出现生殖器官萎缩、闭经,再过一段时间出现甲状腺功能低下及肾上腺皮质功能低下,皮肤色素脱失,阴毛、腋毛、眉毛脱落,进而表现为全身萎缩与老化。

图15-1 Sheenan综合征之垂体
图示垂体前叶坏死
(二)Simond综合征
Simond综合征也是垂体前叶全部激素分泌障碍的一种综合征,呈慢性经过,病程可达30~40年,以出现恶病质和过早衰老为特征。各种激素分泌低下出现的早晚不同,大体按以下顺序出现:GH、FSH/LH、TSH、ACTH、PRL。Simond综合征的病因多为无功能性垂体腺瘤,此外也可以是炎症、循环障碍等引起垂体大面积坏死所致。其他内分泌器官在形态上将先后出现腺体萎缩、间质纤维组织增生及淋巴细胞浸润。
(三)垂体性侏儒症
垂体性侏儒症(pituitary drawfism)是在青春前期的儿童垂体前叶发育障碍或各种病因破坏垂体,引起GH分泌低下所致。表现为骨骼发育障碍,身材矮小,身体各部比例保持儿童期状态。皮肤及颜面出现老人状皱纹,智力发育正常。常伴有一定程度的性腺、甲状腺及肾上腺发育障碍。
四、垂体肿瘤
(一)垂体腺瘤
垂体腺瘤是垂体前叶腺细胞形成的良性肿瘤,占颅内肿瘤10%~15%,临床有特殊表现:①功能性垂体腺瘤分泌过多的某种激素,表现为有关功能亢进,但晚期可由于肿瘤过大压迫血管,引起大面积坏死而转为功能低下;②压迫正常垂体组织使其激素分泌障碍,表现为功能低下;③当直径超过1cm时,将使蝶鞍扩大,直径超过2cm时常向鞍上、蝶窦伸展,压迫视交叉及视神经,引起同侧偏盲或其他视野缺失及其他神经系统症状。
垂体腺瘤生长缓慢,发现时大小不一、小者直径仅数毫米,大者可达10cm,由于CT及核磁共振等影像诊断技术的应用,一般都能早期诊断治疗,巨大腺瘤已十分罕见。肿瘤多数有包膜,少数微小腺瘤可无包膜,不易与局灶性增生区别。肿瘤一般柔软,灰白或微红,有时可见灶状缺血性坏死或出血。镜下瘤细胞呈圆形、多角形,可大可小,在同一肿瘤内大小比较均匀。细胞排列成团状、条索状、片状或乳头状,仅有少量间质及毛细血管将其分隔,少数呈弥漫散在排列。根据瘤细胞染色不同,过去分为以下3种:①嫌色性细胞腺瘤(chromophobe adenoma),约占垂体腺瘤的2/3(图15-2);②嗜酸性细胞腺瘤(acidophile adenoma),约占1/3;③其余为嗜酸性细胞腺瘤(basophile adenoma)。但这种分类不能准确地反映其功能,如嗜酸性细胞腺瘤有的分泌GH,有的分泌PRL,而嫌色细胞腺瘤大部份是有功能的,现在使用特异性抗体及免疫组化技术,从功能上加以鉴别,提出了以下新的分类。
图15-2 垂体嫌色细胞腺瘤
1.生长激素细胞腺瘤(somatotroph adenoma)约占垂体腺瘤的1/4,由于发现常较晚,因而体积较大,在电镜下胞浆中可见直径平均500nm的分泌颗粒,该腺瘤约一半有致密的分泌颗粒,免疫组化染色强阳性,HE切片中胞浆嗜酸性,血中GH水平增高,临床表现为巨人症或肢端肥大症;另一半只有稀疏的分泌颗粒,免疫组化呈弱阳性反应,HE切片胞浆弱嗜酸性或嫌色性,并不分泌过多的GH,因正常垂体组织受其压迫,临床表现为垂体前叶功能低下。
2.催乳素细胞腺瘤(lactotroph adenoma)为垂体腺瘤中最多的一种,约占30%,电镜下胞浆中多为稀疏的小分泌颗粒(图15-3),血中PRL水平增高,在女性早期能出现溢乳-闭经综合征,故发现时肿瘤较小。在男性及老年妇女,催乳素过高血症的症状不明显,因而发现时肿瘤较大。
图15-3 催乳素细胞腺瘤(电镜)
胞浆内可见稀疏的分泌颗粒
3.促肾上腺皮质激素细胞瘤(corticotroph adenoma) 约占垂体腺瘤的15%,组织学上细胞常呈嗜碱性,并呈PAS阳性反应。电镜下分泌颗粒多少不一,大小不等,平均直径约300nm。免疫组化显示ACTH阳性,临床有一半病人表现Cushing综合征,另一半却无该激素功能异常表现,其原因可能是该瘤细胞只合成ACTH的前身,即proopiomelanocortin(POMC)。POMC在细胞内未能分解为ACTH、内腓肽及β-促脂素,因而不具有激素效应。
4.促性腺激素细胞瘤(gonadotroph adenoma)约占5%,光镜下多为嫌色细胞瘤,分泌颗粒显示FSH或FSH及LH,临床性激素功能障碍的症状不明显,多半由于压迫症状才引起注意。
5.促甲状腺细胞腺瘤(thyrotroph adenoma)仅占1%。
6.多种激素细胞腺瘤(plurihormonal adenoma)约占12%~15%,多数为GH细胞及PRL细胞混合腺瘤,有的呈TSH细胞与上述两种细胞分别或共同混合在一起的腺瘤,镜下细胞染色呈多样化。
7.无功能性细胞腺瘤(null cell adenoma)约占25%,镜下为嫌色细胞瘤,免疫组化显示多数细胞激素为阴性,少数细胞可含FSH、TSH及ACTH等,可引起垂体前叶功能低下。
(二)颅咽管瘤
颅咽管瘤(craniopharyngioma)约占颅内肿瘤的1.8%~5.4%,是胚胎期颅咽囊残留上皮发生的肿瘤,肿瘤有的位于蝶鞍内,也可位于蝶鞍外沿颅咽管的各部位。肿瘤大小不一,从小豆大到拳大,瘤体为实性或囊性(单囊或多囊),囊内有暗褐色液体。镜下与造釉细胞瘤相似,瘤细胞排列成巢,细胞巢的周边有1~2层柱状细胞,稍内为棘细胞,中心部逐渐变成星状细胞。细胞巢中心部常有坏死,有胆固醇结晶及钙盐沉着,或液化成囊腔。囊性肿瘤的囊壁由鳞状上皮构成。肿瘤压迫垂体或下丘脑,可引起垂体功能低下;压迫第三脑室可引起脑积水;压迫视神经可引起视野缺失。
第二节 甲状腺疾病
一、非毒性甲状腺肿
非毒性甲状腺肿(nontoxic goiter)亦称单纯性甲状腺肿,是由于甲状腺素分泌不足,促使TSH分泌增多引起的甲状腺肿大。根据地理分布可分为地方性(endemic)和散发性(sporadic)两种。地方性甲状腺肿以远离海岸的内陆山区和半山区多见,人群中约有10%以上的人患有该病,其发病人数是散发性的10倍以上。
祖国医学称甲状腺肿为瘿,隋朝名医巢元方很早就指出瘿瘤的发生与水质有关,晋代(公元3~4世经)就已经使用含碘高的海藻浸酒治疗,在欧洲到1170年才使用海藻灰疗法。
【病因和发病机制】
地方性甲状腺肿的主要病因是缺碘,由于饮水及土壤中缺碘,人体碘摄入不足,导致甲状腺素的合成减少,出现轻度的甲状腺功能低下,通过反馈机制使垂体TSH分泌增多,使甲状腺滤泡上皮细胞增生肥大,因而甲状腺肿大,同时摄取碘的功能增强,提高甲状腺合成分泌甲状腺素的能力,使血中甲状腺素恢复到正常水平,这时增生的上皮逐渐复旧到正常。如果长期持续缺碘,一方面滤泡上皮持续增生,另一方面所合成的甲状腺球蛋白不能充分碘化,不能被上皮细胞吸收利用,从而堆积在滤泡内,使滤泡腔显著扩大,这样使甲状腺进一步肿大。
机体对碘或甲状腺素需求量的增加(例如青春期、妊娠期、授乳期),使机体内甲状腺素相对缺乏,也可招致甲状腺肿。此外有些物质可使甲状腺素合成过程的某个环节发生障碍,也是引起甲状腺肿的附加因素。如长期摄入大量钙,不仅影响碘在肠的吸收,还能使滤泡上皮的细胞膜钙离子增加,抑制甲状腺素的分泌。氟、硼、硅也有类似作用。某些食品如卷心菜、甘蓝、芹菜中含有硫氰酸盐或有机氯酸盐,能够妨碍碘向甲状腺集聚,硫脲能影响一碘酪氨酸向二碘酪氨酸转化,磺胺类药能妨碍酪氨酸的缩合等。
散发性甲状腺肿在女性显著多于男性,其病因不太清楚,除上述因素外,有的与遗传因素有关。
【病变】
按其发展过程,分为3个时期。
1.增生期甲状腺呈弥漫性肿大,表面光滑。镜下滤泡上皮增生肥大,呈立方形或柱状,保持小滤泡新生,胶质含量少,间质充血。甲状腺功能无明显变化。此期可称为弥漫性增生性甲状腺肿。
2.胶质贮积期长期缺碘使滤泡上皮反复增生、复旧,少数滤泡上皮仍呈现增生肥大,保持小型滤泡增生状态,但大部份滤泡显著扩大,内积多量浓厚的胶质,上皮细胞受压变扁平(图15-4)。肉眼见甲状腺弥漫肿大,可达200~300g(正常20~40g),表面光滑,无结节形成,质地较软,切面呈淡褐色,半透明胶冻状(图15-5)。此期可称为弥漫性胶样甲状腺肿(diffuse colloid goiter)。
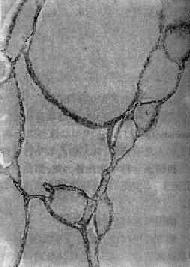
图15-4 地方性甲状腺肿
甲状腺滤泡显著扩大,腔内充满均匀的胶质,上皮细胞变扁平
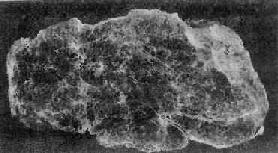
图15-5 弥漫性地方性甲状腺肿 (大体切面)
3.结节期随着病程的发展,由于甲状腺内不同部分滤泡上皮增生与复旧变化不一致,乃形成不规则的结节。镜下与上一期基本相同,只是有的滤泡过度扩大,直径可达300~400μm以上,使滤泡大小差别更大,有的地方亦有滤泡上皮增生,有的增生呈乳头状,可发生癌变。肉眼观甲状腺更加肿大,有许多结节,数量及大小不一,大者直径可达数厘米,结节境界清楚,但无包膜或包膜不完整(图15-6),这是和腺瘤明显不同之处。常发生出血坏死及囊性变,出血和坏死灶可被机化而导致纤维化。
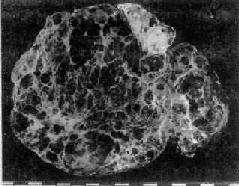
图15-6 结节性地方性甲状腺肿 (大体切面)
肿大的甲状腺可向胸骨下伸延,患者可有颈部压迫感和吞咽困难。甲状腺功能一般仍无明显变化,但少数可有毒性甲状腺肿的症状。
二、毒性甲状腺肿
毒性甲状腺肿(toxic goiter)是具有甲状腺毒症的甲状腺肿。甲状腺毒症(thyrotoxicosis)是由于血中甲状腺素过多作用于全身组织所引起的综合征,其原因:①90%为甲状腺功能亢进,即甲状腺素的合成及分泌增多,如毒性甲状腺肿、毒性腺瘤、毒性结节性甲状腺肿;②甲状腺素释放增多,如某些类型的甲状腺炎;③极少数情况见于垂体促甲状腺细胞腺瘤或下丘脑促甲状腺释放激素的增多,引起继发性甲状腺功能亢进。
毒性甲状腺肿患者年龄常在30~40岁,女性发病比男性高4倍或更高。临床主要表现为甲状腺肿大,甲状腺功能亢进引起的代谢增高、心悸、多汗、多食、消瘦等症状,约有1/3伴有眼球突出,故又称为突眼性甲状腺肿(exophthalmic goiter)。
【病因和发病机制】
病因虽不太清楚,但已有以下证据说明本病为自身免疫性疾病:①与桥本甲状腺炎有许多相似之处,如血中球蛋白增高,并有多种抗甲状腺抗体;常与其他自身免疫性疾病如重症肌无力、血小板减少性紫癜、溶血性贫血等合并发生;②在诸多的抗各种甲状腺成分的抗体中,最重要的是能与TSH受体结合的自身抗体。因为具有类似TSH的作用,其中可分两种,一种是能促进甲状腺素分泌的甲状腺刺激免疫球蛋白(thyroid-stimulating immunoglobulins,TSI),另一种是促进滤泡上皮生长的甲状腺生长刺激免疫球蛋白(thyroid growth-stimulating immunoglobulins,TGI);③本病有家族性素质,在西方已证明在患者及其亲属中HLA-DR3分布频率高,提示有遗传基因素质。有人推测HLA-DR3人群中抑制性T细胞功能是有基因缺陷的,因而辅助性T细胞增强,使自身免疫抗体生成增多。
【病变】
肉眼观,甲状腺对称性弥漫肿大,一般为正常的2~4倍,质较软,切面灰红,胶质含量少。镜下,以滤泡增生为主要特征,滤泡大小不等,以小型滤泡为主。小型滤泡上皮呈立方形,大型滤泡上皮多为高柱状,常向腔内形成乳头状突起。滤泡腔内胶质少而稀薄,胶质的周边部即靠近上皮处出现大小不等的空泡,有的滤泡内甚至不见胶质。间质中血管丰富,显著充血,有多量淋巴细胞浸润并有淋巴滤泡形成(图15-7)。经碘治疗的病例,由于碘能阻断含甲状腺素胶质的分解和促进胶质的储存,故胶质增多变浓,上皮增生受抑制,间质充血减轻,淋巴细胞也减少。与此相反,经硫脲嘧啶等阻断甲状腺素合成的药物治疗者,由于血中TSH代偿性增加,故滤泡增生更明显,上皮呈高柱状,胶质更稀少甚至消失。
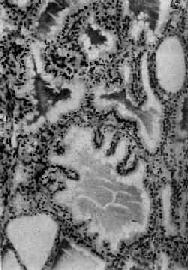
图15-7 毒性甲状腺肿
滤泡增生,有的扩大,上皮呈立方或高柱状,并乳头状突起。滤泡腔内胶质稀薄,周边有大小不等空泡
除甲状腺病变外,全身淋巴组织增生,胸腺肥大和脾肿大;心脏肥大、扩大,心肌可有灶状坏死及纤维化;肝细胞脂肪变性,空泡变性,甚至可有坏死和纤维增生。部份病例有眼球突出,其原因是眼球外肌水肿及淋巴细胞浸润;球后脂肪纤维组织增生,淋巴细胞浸润及大量氨基多糖积聚而形成的粘液水肿,目前认为系自家免疫反应所引起。
三、甲状腺功能低下
甲状腺功能低下(hypothyroidism)是甲状腺素分泌缺乏或不足而出现的综合征,其病因包括:①甲状腺实质性病变,如甲状腺炎,外科手术或放射性同位素治疗造成的腺组织破坏过多,发育异常等;②甲状腺素合成障碍,如长期缺碘、长期抗甲状腺药物治疗、先天性甲状腺素合成障碍、可能由于一种自身抗体(TSH受体阻断抗体)引起的特发性甲状腺功能低下等;③垂体或下丘脑病变。根据发病年龄不同可分为克汀病及粘液水肿。
(一)克汀病
克汀病(cretinism)又称呆小症,是新生儿或幼儿时期甲状腺功能低下的表现,多见于地方性甲状腺肿病区。主要原因是缺碘,在胎儿时期,母亲通过胎盘提供的甲状腺素不足,而胎儿甲状腺及出生之后本身也不能合成足够的激素。散发病例多由于先天性甲状腺素合成障碍。
主要表现为大脑发育不全,智力低下,因为甲状腺素对胎儿及新生儿的脑发育特别重要。此外尚有骨形成及成熟障碍,表现为骨化中心出现延迟,骨骺化骨也延迟,致四肢短小,形成侏儒。头颅较大,鼻根宽且扁平,呈马鞍状,眼窝宽,加上表情痴呆,呈特有的愚钝颜貌。应该指出,在出生后数月内不易察觉智力低下及骨骼发育障碍,而这时又正是脑发育的关键时期,到症状出现再给甲状腺素治疗已无济于事,因此出生后应及早查血,如果T4、T3降低及TSH增高,可确定为甲状腺功能低下。
(二)粘液水肿
粘液水肿(myxedema)是少年及成年人甲状腺功能低下的表现,患者基础代谢显著低下并由此带来各器官功能降低,组织间隙中有大量氨基多糖(透明质酸、硫酸软骨素)沉积而引起粘液水肿,可能是由于该物质分解减慢所致。患者开始表现为怕冷、嗜睡、女性患者有月经不规则,以后动作、说话及思维均减慢,出现粘液水肿。皮肤发凉、粗糙,手足背部及颜面尤其是眼睑苍白浮肿。氨基多糖沉着在声带导致声音嘶哑,沉着在心肌可引起心室扩张,沉着在肠管引起肠蠕动减慢及便秘等。
四、甲状腺炎
甲状腺炎可分为急性、亚急性、慢性三种。急性甲状腺炎为细菌感染引起的急性间质炎或化脓性炎,由于甲状腺对细菌感染抵抗力强,故很少见。亚急性及慢性甲状腺炎是独立的具有特征性病变的疾病。
(一)亚急性甲状腺炎
亚急性甲状腺炎(subacute thyroiditis)又称肉芽肿性或巨细胞性甲状腺炎,一般认为病因是病毒感染,具有发热等病毒感染症状,曾分离出腮腺炎、麻疹、流感病毒,甲状腺出现疼痛性结节,病程为6周到半年,然后自愈。本病女性多于男性,多在30岁左右发病。
肉眼观,甲状腺呈不均匀轻度肿大,质硬,常与周围粘连,切面可见灰白色坏死或纤维化病灶。镜下可见分布不规则的滤泡坏死破裂病灶,其周围有急性、亚急性炎症,以后形成类似结核结节的肉芽肿。肉芽肿中心为不规则的胶质碎块伴有异物巨细胞反应,周围有巨噬细胞及淋巴细胞。以后肉芽肿纤维化,残留少量淋巴细胞浸润。本病初期,由于滤泡破坏甲状腺素释放增多,可出现甲状腺毒症;晚期如果甲状腺有严重的破坏乃至纤维化,可出现甲状腺功能低下。
(二)慢性甲状腺炎
1.慢性淋巴细胞性甲状腺炎(chronic lymphocytic thyroiditis)亦称桥本甲状腺炎,为自身免疫病。患者甲状腺肿大,功能减退。甲状腺结构为大量淋巴细胞、巨噬细胞所取代,滤泡萎缩,结缔组织增生。本病基本缺陷是抗原特异性T抑制细胞减少,致细胞毒性T细胞得以攻击破坏滤泡细胞,且TH细胞参与B细胞形成自身抗体,引起自身免疫反应。
2.纤维性甲状腺炎(fibrous thyroiditis)又称Riedel甲状腺肿,甚少见,主要发生在中年妇女,病因不明。病变多从一侧开始,甲状腺甚硬,表面略呈结节状,与周围明显粘连,切面灰白。镜下,甲状腺滤泡明显萎缩,纤维组织明显增生和玻璃样变,有少量淋巴细胞浸润(图15-8)。临床常有甲状腺功能低下。
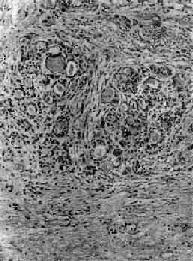
图15-8 纤维性甲状腺炎
甲状腺滤泡萎缩,纤维组织明显增生,有少量淋巴细胞浸润
五、甲状腺肿瘤
(一)甲状腺腺瘤
甲状腺腺瘤(thyroid adenoma)是常见的甲状腺良性肿瘤,多见于青、中年妇女,出现功能亢进者不过1%。肿瘤绝大多数为单发,大小从直径数毫米到3~5cm。肿瘤中心有时可见囊性变、纤维化或钙化。借助以下特点可与结节性胶样甲状腺肿中的结节相区别:有完整的包膜,压迫周围组织,瘤内组织结构比较一致,其形态与周围甲状腺组织不同。病理组织学上可分为以下两种。
1.滤泡性腺瘤(follicular adenoma)根据滤泡分化程度,又可分为以下几种亚型:①胚胎性腺瘤(embryonal adenoma),瘤细胞小,排列成条索状或小片状,有少量不完整的滤泡状腺腔散在,有较多呈水肿的疏松纤维间质;②胎儿型腺瘤(fetal adenoma),由许多小滤泡构成,上皮细胞为小立方形,滤泡腔内多不含胶质,与胎儿甲状腺组织相似(图15-9)。间质较丰富,呈水肿或粘液变性,此型易发生囊性变或出血;③单纯型腺瘤(simple adenoma),由与正常甲状腺相似的滤泡构成,间质较少;④胶样腺瘤(colloid adenoma),滤泡较大,充满胶质,间质少;⑤嗜酸性细胞腺瘤(acidophilic cell adenoma,亦称Hürthle cell adenoma),瘤细胞大而多角形,核小,胞浆丰富,有嗜酸性颗粒,排列成索状或巢状,也可形成不完整的滤泡腔。本瘤较少见。
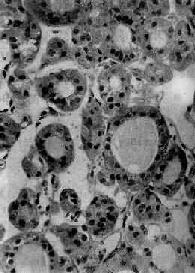
图15-9 胎儿型甲状腺腺瘤
2.乳头状腺瘤(papillary adenoma)滤泡上皮细胞排列成单层,呈乳头状向腺腔内突出,滤泡常形成大囊腔,故亦称囊性乳头状瘤。间质少,肿瘤常并发出血、坏死及纤维化。
(二)甲状腺癌
甲状腺癌(carcinoma of thyroid)在不同地区发病率差别很大,虽然本病恶性程度不同,与其他器官癌相比,发展较缓慢。值得注意的是,有的原发灶很小,临床上常首先发现转移病灶。主要有以下4种类型。
1.乳头状腺癌(papillary adenocarcinoma)占甲状腺癌的40%~60%,青少年女性多见,生长较慢,肉眼发现时多为1~2cm的圆形肿块,无包膜,少数有不完整的包膜,以后逐渐向周围浸润。切面灰色或灰棕色,质地有的较软有的软硬。镜下,癌细胞围绕一纤维血管中心轴呈乳头状排列,乳头分支较多(图15-10)。癌细胞立方形或矮柱状,其特点是核染色质少,呈透明或毛玻璃样,无核仁。可伴有单纯型甲状腺滤泡,间质中常有砂粒体出现。本癌发现时约50%已有颈部淋巴结转移,有时原发灶后于转移灶发现,有的原发灶甚至小到难以觉察的程度。此癌恶性程度低,5年存活率达75%。
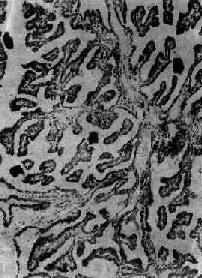
图15-10 甲状腺乳头状腺癌
2.滤泡性腺癌(follicular adenocarcinoma)占甲状腺癌的10%~15%,多见于50岁以上女性。早期即可出现血行转移,原发灶切除后5年存活率为30%~40%。肉眼观,肿瘤灰白色,有的为结节状,有不完整包膜,貌似腺瘤;有的广泛浸润于甲状腺内,进而侵犯气管壁、颈部血管、肌肉及喉返神经。镜下见不同分化程度的滤泡,分化良好者,滤泡结构较规整,细胞异型性亦较低,不易与腺瘤区别(图15-11),须注意包膜或血管是否有瘤细胞浸润来加以鉴别。分化不良者,滤泡少,滤泡形态不整,有的呈实性细胞巢,细胞异型性较明显,核分裂像多见。少数情况下本癌主要由嗜酸性细胞构成,故亦称嗜酸性细胞癌。
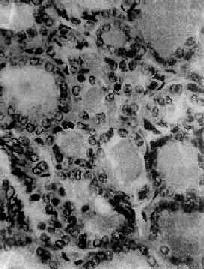
图15-11 甲状腺滤泡性腺癌
3.未分化癌(undifferentiated carcinoma)约占甲状腺癌15%,恶性度高,生长快,早期即可向周围组织浸润并发生转移。患者多在50岁以上,无男女差别。肉眼观,切面灰白色,常有出血、坏死。根据组织形态可分为小细胞型、巨细胞型和梭形细胞型。小细胞型癌由小圆形细胞构成,呈弥漫分布,与恶性淋巴瘤颇相似,用免疫组化鉴别,如瘤细胞显示角蛋白(Keratin)或癌胚抗原(CEA),则可确定其来源于上皮组织。巨细胞型癌预后最差,镜下癌细胞大小不一,形态各异,常有巨核细胞及多核巨细胞(图15-12)。
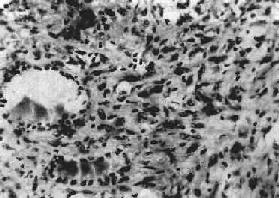
图15-12 甲状腺未分化癌
癌细胞异型性明显,可见巨核细胞
4.髓样癌(medullary carcinoma)是从滤泡旁细胞(亦称C细胞)发生的癌,占甲状腺癌的5%,有的具有家族性,发病年龄在30岁左右,散发病例年龄多在50岁以上。恶性程度不一,平均存活6.6年。90%肿瘤分泌降钙素,有的还同时分泌CEA、生长抑素、前列腺素及其他多种激素和物质,故血中该激素水平增高,表现为典型的多发性内分泌腺瘤。
肉眼观,散发病例开始多为单个肿块,而家族性病例常为多中心性。肿瘤呈黄褐色,较软,境界清晰故乍看似有包膜。镜下瘤细胞为圆形、多角形或梭形小细胞,排列成簇状、索状,偶见小滤泡形成。间质比较丰富,常有淀粉样物质和钙盐沉着。电镜下,瘤细胞胞浆内有直径100~250mm的神经内分泌颗粒。
第三节 甲状旁腺疾病
一、甲状旁腺功能亢进及甲状旁腺增生症
甲状旁腺功能亢进(hyperparathyroidism)分为原发性及继发性两种。
原发性甲状旁腺功能亢进是由于甲状旁腺疾病时分泌过多的甲状旁腺素所致。临床表现为血钙增高及无机磷降低。血钙增高的机制是:①通过破骨细胞的作用引起骨组绢脱钙;②肾小管增加钙的吸收;③胃肠增加钙的吸收。引起本症的主要疾病为甲状旁腺腺瘤与原发性甲状旁腺增生症,其次为甲状旁腺腺癌。
原发性甲状旁腺增生症(primary hyperplasia of parathyroid)的病因不明,四个甲状旁腺几乎都增生,重量可达正常的数百倍,然而增生并非均匀分布,一般以上部甲状旁腺增生较显著。肿大的甲状旁腺由大小不一的红褐色结节组成。镜下,增生的细胞以主细胞为主,排列成条索状;有时以水样透明细胞为主,常呈腺泡状或腺腔排列。
继发性甲状旁腺功能亢进是由于持续性低血钙症引起的继发性甲状旁腺增生症所致,是一种代偿性变化,最常见于慢性肾功能衰竭,也可见于佝偻病、骨软化病、骨髓瘤等。临床上血钙低而血磷高。此时全部甲状旁腺增生肿大,亦可超过正常的数百倍,下部腺体常较上部肿大明显。镜下,增生的细胞主要是主细胞,亦有水样透明细胞和嗜酸性细胞混杂。由于甲状旁腺的代偿作用,血钙可升高到接近正常水平。
二、甲状旁腺肿瘤
甲状旁腺肿瘤大多为单发性,2个以上的多发性腺瘤仅占1%~4%。好发于下部的甲状旁腺,多见于40~60岁,女性较多。重量0.1~5g,有完整包膜,红褐色,质软,光滑,表面可见小的结节。镜下主要为主细胞,呈弥漫性或条索状排列,有时形成腺腔或滤泡状,有时也可见灶状分布的水样透明细胞及嗜酸性细胞,间质甚少。与包膜外正常甲状旁腺的组织像明显不同是与原发性增生症的重要区别。常伴有甲状旁腺功能亢进。
甲状旁腺癌甚少见,其中部分为功能性,可致甲状旁腺功能亢进,好发于30~40岁。肿瘤生长较慢,重量可达10g,灰白色,较硬,向周围浸润。镜下,癌细胞较一致,核分裂像较多,细胞呈条索状排列。约1/3病例可见颈部淋巴结转移,偶尔也可有远处血行转移。
第四节 肾上腺疾病
一、肾上腺皮质功能亢进
肾上腺皮质分泌糖皮质激素(在人类主要是皮质醇)、盐皮质激素及肾上腺雄激素。肾上腺皮持功能亢进(hyperadrenalism)根据何种激素分泌过多可分为不同的综合征,以下两种较多见。
(一)Cushing综合征
Cushing综合征时,糖皮质激素长期分泌过多,促进蛋白异化,继发脂肪沉着。表现为满月脸、向心性肥胖、皮肤变薄并出现紫纹、多毛、痤疮、高血压、糖耐量降低、月经失调及性功能减退、骨质疏松、肌肉无力等。其病因有以下几种。
1.垂体性Cushing综合征 主要由垂体ACTH细胞腺瘤所引起,少数由于下丘脑异常分泌过多的促皮质释放因子(CRF)所致。血清中ACTH增高,双侧肾上腺呈弥漫性中度肥大,重量可达20g(正常8g左右),切面皮质厚度可超过2mm,呈脑回状。镜下主要是网状带及束状带细胞增生(图15-13、图15-14)。
图15-13 肾上腺皮质弥漫增生(大体)
肾上腺肥大,皮质弥漫性增厚,切面呈脑回状
图15-14 肾上腺皮质弥漫增生
图15-13之镜下观,肾上腺皮质束状带弥漫性增厚
2.异位分泌ACTH或CRF肿瘤肾上腺变化与上述同。
3.肾上腺皮质结节性增生(adrenocortical nodular hyperplasia)其原因不明,有的呈家族性。双侧肾上腺明显肥大,重量可超过50g,在弥漫增生的基础上又有许多增生的结节,大小不等,直径从数毫米至2.5cm。镜下,弥漫增生者主为网状带及束状带细胞,而结节内多为束状带细胞,常见多量脂褐素,致结节呈棕褐色。患者血清ACTH水平下降。
4.功能性肾上腺肿瘤 除肿瘤变化外,血清中ACTH减少,致使肾上腺非肿瘤部分萎缩。
5.长期使用糖皮质激素类药物 例如地塞米松(dexamethasone),由于反馈抑制垂体前叶释放ACTH,故血清中ACTH等减少,双侧肾上腺皮质萎缩。
(二)醛固酮增多症
原发性醛固酮增多症(primary aldosteronism)是肾上腺皮质增生的细胞分泌过多的醛固酮所致,引起高血钠症、低血钾症及高血压。本症血清中肾素降低,这是由于钠潴留使血容量增多,抑制了肾素的释放。本病80%是由于功能性肾上腺肿瘤引起,其余为原因不明的两侧肾上腺皮质增生等,这种增生常呈弥漫性,有时也呈结节状,镜下主要为球状带细胞增生,有时也混杂些束状带细胞。
继发性醛固酮增多症是由于各种疾病造成肾素分泌增多所致,肾素可使血浆中的血管紧张素原转变为血管肾张素,后者刺激球状带细胞使醛固酮的分泌增多。
二、肾上腺皮质功能低下
(一)急性肾上腺皮质功能低下症
急性肾上腺皮质功能低下症(acute adrenocortical insufficiency)的病因主要有①皮质大片出血、双侧肾上腺静脉血栓形成,多为败血症的合并症,可能由于毒素损伤了血管,或者由于DIC所致;②在慢性肾上腺皮质功能低下的基础上,由于重症感染、外伤引起了应激反应;③长期皮质激素治疗后突然停药。临床出现血压下降、休克、昏迷等症状,严重时可致死亡。
(二)慢性肾上腺皮质功能低下症
本病又称Addison病,乃由两侧肾上腺皮质严重破坏所致,当破坏超过90%时才出现临床症状,否则为亚临床型。发病呈隐匿性,主要症状是皮肤和粘膜以及瘢痕处的黑色素沉着增加,这是由于肾上腺皮质激素减少,促进垂体ACTH及β-LPH分泌增加,而ACTH及β-LPH在氨基酸排列顺序上有相当一部份与α或β黑色素细胞刺激素(α-MSH、β-MSH)氨基酸顺序相同,故有MSH活性(成人垂体不合成MSH),促进黑色素细胞制造黑色素。由于ACTH分泌不足而导致的继发性肾上腺皮质功能低下时无黑色素沉着,可与本症相鉴别。此外还有低血糖、低血压、肌力低下、易疲劳、食欲不振、体重减轻等症状。
引起本病的病因主要有双侧肾上腺结核(图15-15)和特发性肾上腺萎缩,偶尔也可因转移癌引起。特发性肾上腺萎缩(idiopathic adrenal atrophy)是自家免疫性炎症,故又称自家免疫性肾上腺炎(autoimmue adrenalitis),多见于青年,女性显著多于男性。病人血中常有抗肾上腺皮质细胞线粒体或微粒体抗体,常与其他自家免疫性疾病合并。双侧肾上腺高度萎缩,共重2.5g以下;皮质菲薄,镜下除皮质萎缩外,有大量淋巴细胞和浆细胞浸润。
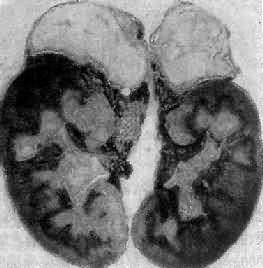
图15-15 双侧肾上腺结核
三、肾上腺肿瘤
(一)肾上腺皮质腺瘤
肾上腺皮质腺瘤与局灶性结节性增生的病变相似,两者可以并发。腺瘤通常是单侧单发性,并有薄层包膜,对周围组织有压迫现象,为鉴别的主要点。大小直径为1~5cm,切面黄色,有时呈红褐色,镜下多为类似束状带的泡沫状透明细胞,含有丰富类脂质,有时由类脂含量少的嗜酸性细胞构成,或者两种细胞混合存在。瘤细胞排列成团,由含有毛细血管的少量间质分隔(图15-16)。部分腺瘤为功能性,可引起醛固酮增多症或Cushing综合征,在形态上与非功能性腺瘤没有区别。
图15-16 肾上腺皮质腺瘤
上图 下图
(二)肾上腺皮质癌
肾上腺皮质癌(adrenocortical carcinoma)甚少见,一般为功能性,发现时一般比腺瘤大,重量常超过100g,呈浸润性生长,正常肾上腺组织破坏或被淹没,向外侵犯周围脂肪组织甚至该侧肾。小的腺癌可有包膜。切面棕黄色,常见出血、坏死及囊性变。镜下分化差者异型性高,瘤细胞大小不等,并可见怪形核及多核,核分裂像多见(图15-17)。常转移到腹主动脉淋巴结或血行转移到肺、肝等处。分化高者镜下像腺瘤,如果癌体小又有包膜,很难与腺瘤区别,有人认为直径超过3cm者,应多考虑为高分化腺癌。
图15-17 肾上腺皮质癌
(三)肾上腺髓质肿瘤
肾上腺髓质来自神经嵴,原始细胞为交感神经母细胞,以后分化为神经节细胞及嗜铬细胞,因此可形成神经母细胞瘤、神经节细胞瘤及嗜铬细胞瘤(神经母细胞瘤及神经节细胞瘤详见第十六章)。
嗜铬细胞瘤(phenochromocytoma) 80%~90%发生于肾上腺髓质,绝大部分为单侧单发性,偶尔见于双侧,90%为良性,好发于30~50岁。肿瘤细胞可分泌去甲肾上腺素和肾上腺素,以去甲肾上腺素为主,偶尔也分泌多巴胺及其他激素,故临床主要有儿茶酚胺过高的症状,表现为血压增高,多呈间歇性发作,并伴有头痛、发汗、末梢血管收缩、脉搏加快、血糖增高、基础代谢上升等症状。肉眼观,大小不一,平均100g左右,甚至可达2000g,有包膜,切面灰红色、灰褐色,常见出血、坏死、囊性变及坏死灶。镜下嗜铬细胞为大多角形细胞,形成细胞索或细胞巢,可有不同程度的多形性,有时出现巨细胞,瘤细胞胞浆内有大量嗜铬的微细颗粒;电镜下,颗粒有包膜,电子致密度低者含有肾上腺素,而包膜不清楚电子致密度高者含有去甲肾上腺素,用免疫组化方法亦可显示。间质主要是血窦。良、恶性肿瘤在细胞形态方面无截然界限,包膜被侵犯並不能作为恶性的肯定证据,但如有周围组织浸润及转移,则属恶性无疑。
肾上腺外嗜铬细胞瘤多为多发性,位于主动脉两侧副节细胞分布处,常称为副节细胞瘤(paraganglioma)。本瘤好发于10~20岁,多数为多发性内分泌肿瘤的一部分,40%为恶性。
第五节 胰岛疾病
一、糖尿病
糖尿病(diabetes mellitus)是由于胰岛素缺乏或(和)胰岛素的生物效应降低而引起的代谢障碍,为以持续的血糖升高和出现糖尿为主征的常见病,发病率为1%~2%。
【病因和发病机制】
糖尿病依病因可分为原发性及继发性两类。继发性糖尿病是由于炎症、肿瘤、手术等已知疾病造成胰岛广泛破坏,或由于其他内分泌的异常影响胰岛素的分泌所导致的糖尿病。日常所称糖尿病乃指原发性糖尿病,按其病因、发病机制、病变、临床表现及预后的不同可分为以下两种。
(一)胰岛素依赖型糖尿病
胰鸟素依赖型糖尿病(insulin-dependent diabetes mellitus,IDDM)又称Ⅰ型糖尿病,占糖尿病的10%~20%,患者多为青少年,发病时年龄小于20岁,胰岛B细胞明显减少,血中胰岛素明显降低,易合并酮血症甚至昏迷,治疗依赖胰岛素。目前认为其发病是在遗传易感性素质的基础上,胰岛感染了病毒(如腮腺炎病毒、风疹病毒及柯萨奇B4病毒等)或受毒性化学物质(如吡甲硝苯脲等)的影响,使胰岛B细胞损伤,释放出致敏蛋白,引起自身免疫反应(包括细胞免疫及体液免疫),导致胰岛的自身免疫性炎症,进一步引起胰岛B细胞严重破坏。
遗传易感性素质的主要根据是:一卵性双生的一方得病,50%的另一方也得病;与HLA类型有明显关系,在中国此型患者中DR3及DR4的分布频率明显增加,有人统计HLA-DR3或DR4的人群患此病的危险性比其他人高5~7倍。这些人存在免疫缺陷,一方面对病毒的抵抗力降低,另一方面抑制性T细胞的功能低下,易发生自身免疫反应(参阅本章毒性甲状腺肿)。
自身免疫反应的主要根据是:患者早期胰岛中有大量淋巴细胞浸润(胰岛炎),其中包括CD4+T细胞,与Ⅰ型糖尿病动物模型所见一致,从Ⅰ型糖尿病动物中提取的CD4+T细胞转移给正常动物可引发该病;90%患者发病后一年内血中可查出抗胰岛细胞抗体;10%的患者同时患有其他自身免疫性疾病。
(二)非胰岛素依赖型糖尿病
非胰岛素依赖型糖尿病(non-insulin dependent diabetes mellitus,NIDDM)又称Ⅱ型糖尿病,发病年龄多在40岁以上,没有胰岛炎症,胰岛数目正常或轻度减少。血中胰岛素开始不降,甚至增高,无抗胰岛细胞抗体,无其他自身免疫反应的表现。本型虽然也有家族性,一卵性双生同时发病者达90%以上,但未发现与HLA基因有直接联系。其发病机制不如Ⅰ型糖尿病清楚,一般认为是与肥胖有关的胰岛素相对不足及组织对胰岛素不敏感(胰岛素抵抗)所致。
肥胖是本型发生的重要因素,患者85%以上明显肥胖,只要减少进食,降低体重,血糖就可下降,疾病就可得到控制。引起发病有两个重要环节:①胰岛素相对不足及分泌异常。长期高热量食物,刺激胰岛B细胞,引起高胰岛素血症,但与同样肥胖的非糖尿病者相比,血中胰岛素水平较低,因此胰岛素相对不足。此外对葡萄糖等刺激,胰岛素早期呈现延缓反应,说明胰岛B细胞本身也有缺陷。长期过度负荷可使胰岛B细胞衰竭,因此本病晚期可有胰岛素分泌绝对缺乏,不过远比Ⅰ型为轻。②组织胰岛素抵抗,脂肪细胞越大对胰岛素就越不敏感,脂肪细胞及肌细胞的胰岛素受体减少,故对胰岛素反应差,这是高胰岛素血症引起胰岛素受体负调节的表现;此外营养物质过剩的细胞还存在胰岛素受体后缺陷(defect of postreceptor signaling by insulin),使葡萄糖及氨基酸等不能通过细胞膜进入细胞内,事实上营养过剩的细胞(也包括肝细胞及肌细胞)已失去正常处理血液中营养物质的能力。
非肥胖型Ⅱ型糖尿病患者对葡萄糖早期胰岛素反应比肥胖型患者更差,提示胰岛B细胞缺陷更严重,同时组织也呈胰岛素抵抗,其原因不明,可能与基因异常有关。
糖尿病患者在临床上不仅呈糖代谢障碍,也有脂肪及蛋白代谢障碍。胰岛素的不足(绝对或相对)及组织胰岛素抵抗使葡萄糖利用及糖原合成减少,导致高血糖。血糖超过肾阈值则出现尿糖及高渗性利尿(多尿)。这将引起水及电解质的丢失,进一步导致细胞内水减少,故患者出现口渴。由于营养物质得不到利用,患者食欲常增强,而体重却减轻(主要见于Ⅰ型及严重的Ⅱ型糖尿病)。
在胰岛素严重缺乏情况下(见于Ⅰ型糖尿病),蛋白及脂肪分解代谢增强而生成氨基酸及脂肪酸,氨基酸在肝内作为糖异生的原料被利用,而脂肪酸则在肝内氧化生成酮体(乙酰乙酸、β-羟丁酸及丙酮),出现酮血症(ketonemia)及酮尿症(ketonuria),前者可导致糖尿病昏迷。部分患者胰岛A细胞分泌的高血糖素(glucagon)增高,能加速脂肪酸的氧化。
【病理变化】
1.胰岛的病变不同类型的糖尿病及其不同时期,病变差异甚大。Ⅰ型糖尿病早期可见胰岛炎,胰岛内及其周围有大量淋巴细胞浸润,偶见嗜酸性粒细胞。胰岛细胞进行性破坏、消失,胰岛内A细胞相对增多,进而胰岛变小,数目也减少,有的胰岛纤维化;Ⅱ型糖尿病用常规方法检查时,早期几无变化,以后可见胰岛B细胞有所减少。常见变化为胰岛淀粉样变,在B细胞周围及毛细血管间有淀粉样物质沉着(图15-18),该物质可能是胰岛素B链的分解产物。
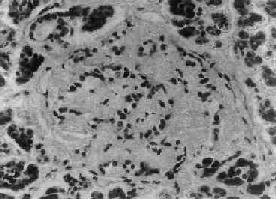
图15-18 糖尿病之胰岛
图示胰岛淀粉样变
2.其他组织变化及合并症
(1)动脉病变:①动脉粥样硬化,比非糖尿病患者出现较早且较严重;②细动脉玻璃样变,表现为基底膜增厚,富于Ⅳ型胶原的物质沉着,由于通透性增高致蛋白质漏出增多,故动脉壁有蛋白质沉积,造成管腔狭窄,引起组织缺血。合并高血压者,此变化更明显。
(2)肾病变:①肾小球硬化,有两种类型,一种是弥漫性肾小球硬化(diffuse glomerulosclerosis),肾小球毛细血管基底膜弥漫增厚,血管系膜细胞增生及基质增多;另一种为结节性肾小球硬化(nodular glomerulosclerosis),其特点是部分系膜轴有多量透明物质沉着,形成结节状,结节外周为毛细血管袢。②动脉硬化及小动脉硬化性肾硬化。③急性和慢性肾盂肾炎,易伴有肾乳头坏死,后者是由于在缺血的基础上对细菌感染更加敏感。④肾近曲小管远端上皮细胞有糖原沉积。
(3)糖尿病性视网膜病。可分两种,一种是背景性视网膜病(background retinopathy),视网膜毛细血管基底膜增厚,小静脉扩张,常有小血管瘤形成,继而有水肿、出血;另一种是由于血管病变造成视网膜缺氧,刺激引起血管新生及纤维组织增生,称为增殖性视网膜病(proliferative retinopathy)。视网膜病变易引起失明。除视网膜病变外,糖尿病易合并白内障。
(4)神经系统病变:周围神经包括运动神经、感觉神经和植物神经都可因血管变化引起缺血性损伤,出现各种症状,如肢体疼痛、麻木、感觉丧失、肌肉麻痹以致足下垂、腕下垂、胃肠及膀胱功能障碍等;脑细胞也可发生广泛变性。
(5)其他器官病变:肝细胞核内糖原沉积;由于高血脂症皮肤可出现黄色瘤结节或斑块。
(6)糖尿病性昏迷:其原因有①酮血症酸中毒;②高血糖引起脱水及高渗透压。
(7)感染:由于代谢障碍及血管病变使组织缺血,极易合并各种感染。
二、胰岛细胞瘤
胰岛细胞瘤(islet-cell tumor)由于其构成细胞不同,所分泌的激素和引起的症状也不同。有一部分肿瘤乃无功能性肿瘤,临床上不出现任何特殊症状。胰岛细胞瘤在HE染色片上不可能区分出细胞的种类,常需借助特殊染色、电镜及免疫组化技术来加以鉴别。
1.B细胞肿瘤 从胰岛B细胞(即B细胞)发生的肿瘤有腺瘤和腺癌;其中一部分能分泌胰岛素,可称之为insulinoma,临床上可有低血糖症状。大约80%是单发性腺瘤,10%为多发性腺瘤,10%是癌。本瘤可见于各个年龄层,但以40~50岁为多见。肉眼观,一般以胰体及胰尾部为多见,有完整包膜,大小为0.5~10cm,切面色微黄。组织学上,瘤细胞似胰岛细胞,呈多角形或柱状,胞浆颗粒状,细胞排列成条索或巢状,有时可出现菊形团排列。周围绕以血窦(图15-19)。间质中常出现淀粉样物质或钙盐沉积。其恶性者称为胰岛细胞癌。但由于肿瘤细胞常可因变性而出现多形性和奇异形核,因而从镜下区分良恶性有时也颇困难,常需根据其浸润性生长和淋巴结、肝转移等才能做出判断。
图15-19 胰岛细胞瘤
2.Zollinger–Ellison综合征本综合征以胰岛细胞瘤、大量胃酸分泌和溃疡病为主征。肿瘤常为多发性,60%是恶性,细胞大小不一,瘤细胞分泌胃泌素,故又称胃泌素瘤(gastrinoma)。在胰岛该瘤细胞来源不明。此瘤可见于胰腺任何部位,也可发生在十二指肠及胃幽门窦部。由于胃泌素的作用,胃酸分泌亢进,导致溃疡病形成。溃疡主要见于十二指肠及胃,但25%可出现在空肠。患者还常有水样腹泻及脂性腹泻。
第六节 APUD系统肿瘤
APUD(amine precursor uptake and decarboxylation)系统是来源于神经嵴的一系列内分泌细胞,弥散在许多器官及内分泌腺体内,能够从细胞外摄取胺的前体,并通过细胞内氨基脱羧酶的作用,使胺前体形成相应的胺(如多巴胺、5-羟色胺等)和多肽激素。现在又把这种细胞称为弥散神经内分泌细胞(diffuse neuroendocrine cells),并已发现40余种之多:在中枢存在于下丘脑-垂体轴和松果体,在周围存在于胃、肠、胰、呼吸系统、泌尿生殖系统、甲状腺、甲状旁腺及肾上腺髓质。如甲状腺有分泌降钙素的C细胞,胃有分泌胃泌素的G细胞、胰岛有分泌胰岛素的B细胞及分泌高血糖素的A细胞、十二指肠有分泌肠促胰肽(secretin)的S细胞和分泌胃性抑制肽(gastric inhibitory peptide)的细胞等等。从这种细胞发生的肿瘤可以统称之为APUD瘤。目前由于免疫组化和免疫细胞化学技术的进步,已能加以鉴别并起用各自的名称,如ACTH瘤、胃泌素瘤、血管活性肠肽瘤(VIPoma)等。
一、多发性内分泌肿瘤
多发性内分泌肿瘤(multiple endocrine neoplasms,MEN)是指在两个以上内分泌腺发生肿瘤或增生,出现多种内分泌功能障碍,有明显的家族遗传性。根据不同组合可分为MEN-Ⅰ(Wermer综合征)、MEN-Ⅱ(Sipple综合征)及MEN-Ⅲ3种类型(表15-1)。
表15-1 MEN分类
| MEN-Ⅰ | MEN-Ⅱ | MEN-Ⅲ | |
| 垂体前叶 | 腺瘤 | ||
| 甲状旁腺 | 增生+++ | 增生++ | |
| 腺瘤+ | |||
| 胰岛 | 增生+ | ||
| 腺瘤++ | |||
| 癌+++ | |||
| 肾上腺 | 皮质增生++ | 嗜铬细胞瘤++ | 嗜铬细胞瘤+++ |
| 甲状腺 | C细胞增生± | 髓样癌+++ | 髓样癌++ |
| 其他 | 粘膜下神经纤维瘤 | ||
| Marfan样体型* | |||
| 染色体基因突变点 | 11q13 | 10 | 不明 |
注:+不常见;+++常见
*Marfan样体型:患者消瘦,四肢及指骨细长,肌肉不发达,张力低,关节伸展过度,脊柱后凸,足外翻,弓形足,漏斗胸等畸形,是结缔组织异常的遗传性疾病。
二、异位产生激素的肿瘤
正常情况下不产生激素的组织或器官发生能分泌激素的肿瘤,称为异位产生激素的肿瘤(ectopic hormone producing tumor)。常分泌的激素有ACTH、抗利尿激素(ADH)、甲状旁腺激素(PTH),降钙素(CT)、绒毛膜促性腺激素(HCG)等。
异位性分泌ACTH的肿瘤,如肺的燕麦细胞癌、腺癌、未分化癌,其次如胰腺癌、支气管类癌、消化道类癌、嗜铬细胞瘤及甲状腺髓样癌等。
异位性分泌ADH的肿瘤,如肺癌(燕麦细胞癌、未分化癌)、胰腺癌;十二指肠肿瘤、前列腺癌等。
异位性分泌PTH的肿瘤,如肺癌(鳞癌)、肾癌、卵巢癌、胰腺癌、肝癌、膀胱癌等。
异位性分泌HCG的肿瘤,如肺癌(大细胞癌,鳞癌),患者多为老年男性,临床上可有乳腺发育症。由肝癌引起者多为男性幼儿,临床上可出现性早熟。
异位性分泌红细胞生成素的肿瘤有肾癌、肺癌、小脑血管母细胞瘤、子宫纤维瘤、嗜铬细胞瘤等。临床上出现红细胞增多症。
第十六章 神经系统疾病
由于神经系统解剖生理学上的某些特殊性,故在病理方面具有和其它实质性器官(如肝、肾)不同的一些特殊规律:①病变定位和功能障碍之间的关系密切,例如一侧大脑额叶前中央回病变可导致对侧肢体偏瘫;②相同的病变发生在不同的部位,可出现不同的综合征及后果,如额叶前皮质区(联络区)的小梗死灶可不产生任何症状,而如发生在延髓则可导致严重后果,甚至致命;③对各种致病因子的病理反应较为刻板,同一种病变可出现在许多不同的疾病中,例如炎症渗出过程往往表现为血管套的形成;④某些解剖生理特征具有双重影响,如颅骨虽具保护作用,但又是引起颅内高压和脑疝的重要因素。
第一节 神经系统对损伤的基本反应
一、神经元与胶质细胞反应
(一)神经元
神经元是中枢神经系统的基本结构和功能单位,由细胞体和胞突(树突、轴突)构成。其数目估计在百亿以上。神经元常见的病变为:
1.中央性Nissl小体溶解(central chromatolysis)这是一种可逆性变化,病因一旦去除,就可恢复正常,如病变继续发展,则可导致细胞的萎缩和死亡。常见的病因有病毒感染(如脊髓灰质炎病毒)、维生素B属缺乏、坏血病和神经元与轴突断离。病变表现为神经细胞肿胀,丧失典型的多极形状而变为圆形,胞核偏位,胞浆中央的Nissl小体崩溃成为细尘状颗粒,进而完全溶解消失,或仅在细胞周边部有少量残余。胞浆着色浅而呈苍白均质状(图16-1)。
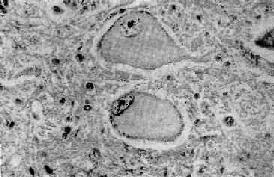
图16-1 中央Nissl小体溶解
神经细胞肿胀,胞核偏位,Nissl小体仅见于细胞周边部
切断实验动物的轴突后,相关的神经细胞即发生典型的中央性Nissl小体溶解,此现象又称轴突反应。此时神经细胞粗面内质网的核蛋白体解聚,成为游离核蛋白体,蛋白质合成加快,因此认为其与轴突再生有关。
2.神经元急性坏死缺血、缺氧、严重急性中毒或感染可引起神经元的死亡。表现为神经细胞核固缩,胞体缩小变形,胞浆Nissl小体消失,呈深伊红色,称为红色神经细胞。如细胞坏死后的酶性分解过程继续发展,则可导致细胞溶解和消失。隐约可见轮廓的死亡细胞称为鬼影细胞(ghost cell)。因缺血引起的神经细胞坏死最常见于大脑皮质的锥体细胞和小脑蒲肯野(Purkinje)细胞(图16-2)。
图16-2 神经细胞缺血性变化
细胞缩小呈三角形,Nissl小体消失,胞核浓缩深染
3.神经元的慢性病变为一组特殊的病变,如单纯性萎缩、神经元纤维的缠结、神经细胞胞浆中出现特殊的包含体(如Lewy小体)等。
4.轴索和髓鞘的变化——Waller变性是指神经纤维被切断后,轴索与神经元胞体断离,其远端和部分近端的轴索及其所属髓鞘发生变性、崩解和被吞噬细胞吞噬的过程。与此同时,受累神经元的胞体发生中央性Nissl小体溶解。除了机械性损伤外,任何其他原因只要能造成神经元胞体与轴索断离者(如循环障碍所致的大脑皮质梗死)均可发生Waller变性,其整个过程包括:①轴索变性,表现为远端轴索肿胀、断裂、崩解、被吞噬细胞吞噬消化;近端的轴索则随后再生并向远端延伸;②髓鞘脱失,髓鞘崩解所形成的脂质和中性脂肪,可被苏丹Ⅲ染成红色。③细胞反应,表现为吞噬细胞反应性增生,吸收崩解产物。周围神经断端远侧Schwann细胞(许旺细胞)反应性增生;而在中枢神经系统则为少突胶质细胞增生,两者均参与再生轴突的重新髓鞘化过程(图16-3)。
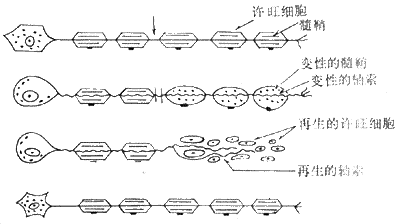
图16-3 Waller变性模式图 (采自Escourolle)
(二)神经胶质细胞
神经胶质细胞包括星形胶质细胞、少突胶质细胞和室管膜细胞,其总数是神经元的5倍,其基本病变概括如下:
1.星形胶质细胞
(1)星形胶质细胞增生:常见的致病原因如缺氧、低血糖、感染、中毒等均能引起脑组织的损伤而导致星形胶质细胞增生。反应性星形胶质细胞增生是脑组织损伤的修补愈合反应,主要表现为纤维型星形胶质细胞增生,最后成为胶质瘢痕,胶质纤维酸性蛋白(GFAP)染色呈强阳性(图16-4)。胶质瘢痕与纤维瘢痕不同之处在于星形胶质细胞并不产生胶原纤维及相应间质蛋白。胶持瘢痕是由星形胶质细胞突起构成,其机械强度不如胶原瘢痕。
图16-4 反应性星形胶质细胞增生(GFAP染色)
左上角为脑毁损病灶
(2)星形胶质细胞肥大:表现为细胞体积增大,胞浆丰富,呈伊红着色,胞核偏位。电镜下可见胞浆中充满线粒体、内质网、空泡、高尔基器、溶酶体和胶质纤维。肥胖星形胶质细胞(gemistocytic astrocyte)多发生在脑的局部缺氧、水肿、梗死、脓肿或肿瘤周围。
2.少突胶质细胞少突胶质细胞的病变常表现为髓鞘的改变,白质营养不良(髓鞘形成障碍)和脱髓鞘疾病如多发性硬化为其代表。此类胶质细胞对各种损害(缺氧、中毒等)所表现的急性肿胀、核周空晕,目前认为可能是一种自溶性变化。此外神经元胞体周围被5个以上少突胶质细胞围绕称为卫星现象(satellitosis),与神经元损害的程度和时间并无明显的关系,其意义尚不清楚。
3.室管膜细胞室管膜细胞覆盖在脑室系统内面,各种致病因素均可引起局部室管膜细胞丢失,随之室管膜下的星形胶质细胞增生,充填缺损,并形成多数小颗粒向脑室突出,称为颗粒性室管膜炎(图16-5)。巨细胞病毒感染时,室管膜细胞中往往可有病毒包含体出现。
图16-5 颗粒性室管膜炎
局部室管膜细胞丢失,增生的胶质细胞隆起,形成小颗粒
(三)小胶质细胞
小胶质细胞属单核吞噬细胞系统,其对损害之反应表现为:①激活形成巨噬细胞,包围、浸润和吞噬坏死神经元,称为噬神经细胞现象(neuronophagia),吞噬后胞浆中常出现大量小脂滴,常规石蜡切片,HE染色呈空泡状,又称格子细胞或泡沫细胞,苏丹染色呈阳性反应;②局灶性增生形成胶质结节,常见于各种炎症,特别是病毒性脑炎;③杆状细胞形成,见于慢性进行性损害(如脑晚期梅毒),表现为细胞增生、胞体变窄、胞突减少并呈双极杆状。
二、常见的并发症
中枢神经系统疾病最常见而重要的并发症为颅内压升高、脑水肿和脑积水,其中脑水肿和脑积水可引起或加重颅内压升高,三者可合并发生,互为因果,后果严重可导致死亡。
(一)颅内压升高及脑疝形成
侧卧位的脑脊液压超过2kPa(正常为0.6~0.8kPa)即为颅内压增高,这是由于颅内容物的容积增加,超过了颅腔所能代偿的极限所致。颅内压增高的主要原因是颅内占位性病变和脑脊液循环阻塞所致的脑积水。常见的占位性病变为脑出血和血肿形成(如创伤、高血压脑出血等),脑梗死,肿瘤,炎症(如脑膜脑炎、脑脓肿等),脑膜出血等。其后果与病变的大小及其增大的速度有关。脑水肿可加重病变的占位性。颅内压升高可区别为3个不同的时期:
1.代偿期通过反应性血管收缩以及脑脊液吸收增加和形成减少,使血容量和脑脊液容量相应减少,颅内空间相对增加,以代偿占位性病变引起的脑容积增加。
2.失代偿期占位性病变和脑水肿使颅内容物容积继续增大,超过颅腔所能容纳的程度,可引起头痛、呕吐、眼底视乳头水肿、意识障碍、血压升高及反应性脉搏变慢和脑疝形成。
3.血管运动麻痹期颅内压严重升高使脑组织灌流压降低,致脑缺氧造成脑组织损害和血管扩张,继而引起血管运动麻痹,加重脑水肿,引起昏迷及并发症,后果严重,可导致死亡。
升高的颅内压可引起脑移位、脑室变形、使部分脑组织嵌入颅脑内的分隔(大脑镰,小脑天幕)和颅骨孔道(如枕骨大孔等)导致脑疝形成(herniation)。常见的脑疝有以下类型。
1.扣带回疝又称大脑镰下疝,是因一侧大脑半球特别是额、顶、颞叶的血肿或肿瘤等占位性病变,引起中线向对侧移位,同侧扣带回从大脑镰的游离边缘向对侧膨出,形成扣带回疝。疝出的扣带回背侧受大脑镰边缘压迫形成压迹,受压处的脑组织发生出血或坏死。此外,大脑前动脉的胼胝体支也可受压引起相应脑组织梗死。大脑冠状切面上可见对侧的侧脑室抬高,第三脑室变形,状如新月(图16-6)。
图16-6 脑疝模式图
1.扣带回疝;2.海马钩回疝;3.小脑扁桃体疝;4.硬膜外血肿
2.小脑天幕疝又称海马钩回疝。位于小脑天幕以上的额叶或颞叶内侧的肿瘤、出血、梗死等病变引起脑组织体积肿大,导致颞叶的海马钩回经小脑天幕孔向下膨出。海马钩回疝可导致以下后果:①同侧动眼神经在穿过小脑天幕裂孔处受压,引起同侧瞳孔一过性缩小,继之散大固定,及同侧眼上视和内视障碍。②中脑及脑干受压后移,可导致意识丧失;导水管变狭,脑脊液循环受阻加剧颅内压的升高;血管牵伸过度,引起中脑和桥脑上部出血梗死,可导致昏迷死亡。③中脑侧移,使对侧中脑的大脑脚抵压于该侧小脑天幕锐利的游离缘上,形成Kernohan切迹。严重时该处脑组织(含锥体索)出血坏死,导致与天幕上原发病变同侧的肢体瘫痪,引起假定位症。④压迫大脑后动脉引起同侧枕叶距状裂脑组织出血性梗死(图16-7)。
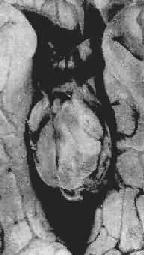
图16-7 海马钩回疝
左海马回内侧肿胀有深切迹(箭头),中脑右移变形,中脑右大脑脚受压,局部坏死出血(Kemohan切迹)
3.小脑扁桃体疝又称枕骨大孔疝。主要由于颅内高压或后颅凹占位性病变将小脑和延髓推向枕骨大孔并向下移位而形成小脑扁桃体疝。疝入枕骨大孔的小脑扁桃体和延髓成圆锥形,其腹侧出现枕骨大孔压迹(图16-8),由于延髓受压,生命中枢及网状结构受损,严重时可引起呼吸变慢甚至骤停,接着心脏停搏而猝死。
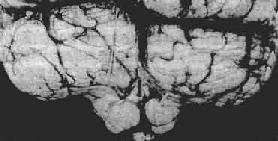
图16-8 小脑扁桃体疝
示小脑切迹,两侧扁桃体疝形成
(二)脑水肿
脑组织中由于液体过多贮积而形成脑水肿(brain edema),这是颅内压升高的一个重要原因。许多病理过程如缺氧、创伤、梗死、炎症、肿瘤、中毒等均可伴发脑水肿。
脑组织易发生水肿与下列解剖生理特点有关:①血脑屏障的存在限制了血浆蛋白通过脑毛细血管的渗透性运动;②脑组织无淋巴管以运走过多的液体。常见脑水肿的类型为:
1.血管源性脑水肿最为常见,是血管通透性增加的结果,当毛细血管内皮细胞受损,血脑屏障发生障碍时,或新生毛细血管尚未建立血脑屏障时(如转移性肿瘤及脑脓肿周围有大量的新生毛细血管),血液中的液体大量渗入细胞外间隙,引起脑水肿。白质水肿较灰质更为明显。此型水肿常见于脑肿瘤、出血、创伤或炎症时。水肿液较富于蛋白质。
2.细胞毒性脑水肿多见于缺血或中毒引起的细胞损害。由于细胞膜的钠-钾依赖性ATP酶失活,细胞内水、钠潴留,引起细胞(神经细胞、胶质细胞、内皮细胞)肿胀,细胞外间隙减小。此型水肿可同样累及灰质和白质。
上述两型水肿常同时存在,尤其在缺血性脑病时更为显著。
脑水肿的肉眼形态为脑体积和重量增加,脑回宽而扁平,脑沟狭窄,白质水肿明显,脑室缩小,严重的脑水肿常同时有脑疝形成。镜下,脑组织疏松,细胞和血管周围空隙变大,白质中的变化较灰质更加明显。电镜下,细胞外间隙增宽,星形胶质细胞足突肿胀(血管源性水肿),或无间隙增宽仅有细胞肿胀(细胞毒性水肿)。
(三)脑积水
脑脊液量增多伴脑室扩张称为脑积水(hydrocephalus)。脑积水发生的主要原因是脑脊液循环的通路被阻断。引起脑脊液循环受阻的原因很多,诸如先天畸形、炎症、外伤、肿瘤、蛛网膜下腔出血等。脑室内通路阻塞引起的脑积水称阻塞性或非交通性脑积水;如脑室内通畅而因蛛网膜颗粒或绒毛吸收脑脊液障碍所致的脑积水称交通性脑积水。此外脉络丛乳头状瘤分泌过多脑脊液也可导致脑积水。
轻度脑积水时,脑室轻度扩张,脑组织呈轻度萎缩。严重脑积水时,脑室高度扩张,脑组织受压萎缩、变薄,脑实质甚至可菲薄如纸,神经组织大部分萎缩而消失(图16-9)。
图16-9 脑积水
侧脑室高度扩张,脑组织受压萎缩变薄
婴幼儿颅骨缝闭合前如发生脑水肿,患儿可出现进行性头颅变大,颅骨缝分开,前囟扩大;颅内压增高较轻,头痛,呕吐,视乳头水肿也出现较晚。由于大脑皮质萎缩,患儿的智力减退,肢体瘫痪。成人脑积水,因颅腔不能增大,颅内压增加的症状发生较早也较严重。
第二节 感染性疾病
中枢神经系统的感染性疾病,按病因分有病毒、细菌、立克次体、螺旋体、真菌、寄生虫等引起的疾病。病原体可通过下列途径入侵中枢神经系统:①血源性感染——脓毒血症,感染性栓子等。②局部扩散——颅骨开放性骨折、乳突炎、中耳炎、鼻窦炎等;③直接感染,创伤或医源性(腰椎穿刺等)感染;④经神经感染,某些病毒发狂犬病病毒可沿周围神经,单纯疱疹病毒可沿嗅神经、三叉神经入侵中枢神经而引起感染。神经系统的免疫部署特点在于:①血脑屏障和血管周围间隙(Virchow-Robin space,V-R间隙)不仅构筑了一条天然防线,而且在一定程度上限制了炎症反应向脑实质伸展;②无固有的淋巴组织和淋巴管,免疫活性T、B细胞均须由周围血液输入。
一、细菌性疾病
(一)脑膜炎
脑膜炎(meningitis)可累有硬脑膜、蛛网膜和软脑膜。硬脑膜炎(pachymeningitis)多继发于颅骨感染。自从抗生素广泛应用以来,此病之发病率已大为减少。软脑膜炎包括蛛网膜和软脑膜炎症,则颇为常见。因此目前脑膜炎实际上是指软脑膜炎(leptomeningitis)而言。脑膜炎绝大部分由病原体引起,由脑膜炎双球菌引起的流行性脑膜炎是其中最主要的类型;少数则由刺激性化学药品(如普鲁卡因、氨甲蝶呤)引起。脑膜炎有3种基本类型:化脓性脑膜炎,淋巴细胞性脑膜炎(多由病毒引起),慢性脑膜炎(可由结核杆菌、梅毒螺旋体、布氏杆菌及真菌引起)。本节重点叙述化脓性脑膜炎。
1.急性化脓性脑膜炎急性化脓性脑膜炎是软脑膜的急性炎症,大量炎性渗出物积聚于蛛网膜下腔。其中流行性脑膜炎多在冬春季流行,其余病原的脑膜炎则多为散发性。
【病因和发病机制】
急性化脓性脑膜炎的致病菌类型随患者之年龄而异。在青少年患者中以脑膜炎双球菌感染为主。该菌存在于病人和带菌者的鼻咽部,借飞沫经呼吸道传染,细菌进入上呼吸道后,大多数只引起局部炎症,成为健康带菌者;仅小部分机体抵抗力低下的患者,细菌可从上呼吸道粘膜侵入血流,并在血液中繁殖,到达脑脊膜后引起脑膜炎。在冬春季可形成流行,称为流行性脑膜炎。
新生儿脑膜炎最常见的病因是大肠杆菌,感染多来自产道。由于体内缺乏能中和病菌的IgM,入侵的大肠杆菌得以繁殖而致病。
流感杆菌脑膜炎多见于3岁以下之婴幼儿。肺炎球菌脑膜炎在幼儿和老年人常见,其中幼儿的脑膜感染多来自中耳炎,而在老人则常为大叶性肺炎的一种并发症。
【病理变化】
肉眼观,脑脊膜血管高度扩张充血,病变严重的区域,蛛网膜下腔充满灰黄色脓性渗出物,覆盖着脑沟脑回,以致结构模糊不清(图16-10),边缘病变较轻的区域,可见脓性渗出物沿血管分布。在渗出物较少的区域,软脑膜往往略带混浊。脓性渗出物可累及大脑凸面矢状窦附近或脑底部视神经交叉及邻近各池。由于炎性渗出物的阻塞,使脑脊液循环发生障碍,可引起不同程度的脑室扩张。
图16-10 化脓性脑膜炎
蛛网膜下腔内有多量脓液堆积以致大部分脑表面的沟回结构不清;脑膜血管高度扩张充血
镜下,蛛网膜血管高度扩张充血,蛛网膜下腔增宽,其中有大量中性粒细胞及纤维蛋白渗出和少量单核细胞、淋巴细胞浸润(图16-11)。用革兰染色,在细胞内外均可找到致病菌。脑膜及脑室附近脑组织小血管周围可见少量中性粒细胞浸润。病变严重者,动、静脉管壁可受累并进而发生脉管炎和血栓形成,从而导致脑实质的出血性梗死。

图16-11 化脓性脑膜炎
蛛网膜下腔充满脓性渗出物,血管扩张充血
【临床病理联系】
急性化脓性脑膜炎在临床上除了发热等感染性全身性症状外,常有一系列神经系统症状,表现为:
(1)颅内压升高症状:头痛,喷射性呕吐,小儿前囟饱满等。这是由于脑血管充血,蛛网膜下腔渗出物堆积,蛛网膜颗粒因脓性渗出物阻塞而影响脑脊液吸收所致,如伴有脑水肿,则颅内压升高更加显著。
(2)脑膜刺激症状:颈项强直。炎症累及脊髓神经根周围的蛛网膜、软脑膜及软脊膜,致使神经根在通过椎间孔处受压,当颈部或背部肌肉运动时可引起疼痛,颈项强直是颈部肌肉对上述情况所发生的一种保护性痉挛状态。在婴幼儿,由于腰背肌肉发生保护性痉挛可引起角弓反张(episthotonus)的体征。此外,Kerning征(屈髋伸膝征)阳性,是由于腰骶节段神经后根受到炎症波及而受压所致,当屈髋伸膝试验时,坐骨神经受到牵引,腰神经根因压痛而呈现阳性体征。
(3)颅神经麻痹:由于基底部脑膜炎累及自该处出颅的Ⅲ,Ⅳ,Ⅴ,Ⅵ,Ⅶ对颅神经,因而引起相应的神经麻痹征。
(4)脑脊液的变化:压力上升,混浊不清,含大量脓细胞,蛋白增多,糖减少,经涂片和培养检查可找到病原体。脑脊液检查是本病诊断的一个重要依据。
结局和并发症
由于及时治疗和抗菌素的应用,大多数患者可痊愈,病死率已由过去70%~90%降低到50%以下。如治疗不当,病变可由急性转为慢性,并可发生以下后遗症:①脑积水:由于脑膜粘连,脑脊液循环障碍所致;②颅神经受损麻痹:如耳聋,视力障碍,斜视、面神经瘫痪等;③脑底脉管炎致管腔阻塞,引起相应部位脑缺血和梗死。
暴发性脑膜炎球菌败血症是暴发型流行性脑脊膜炎的一种类型,多见于儿童。本病起病争骤,主要表现为周围循环衰竭、休克和皮肤大片紫瘢。与此同时,两侧肾上腺严重出血,肾上腺皮质功能衰竭,称为沃-弗(Waterhouse-Friederichsen)综合片,其发生机制主要是大量内毒素释放所引起的弥漫性血管内凝血,患者脑膜变化轻微,病情凶险,一般在起病24小时内死亡。
2.结核性脑膜炎 详见第十八章
(二)脑脓肿
【病因】
脑脓肿的致病菌既往以葡萄球菌,链球菌,肺炎球菌等需氧菌为多见,近年来厌氧菌属之链球菌,无芽胞革兰阴性菌,类杆菌等亦成为常见的致病菌。
感染途径
1.直接蔓延病菌通过颅骨骨折伤口或局部感染灶直接蔓延至脑而引起的脑脓肿,其中常见的局部感染灶为中耳炎、乳突炎和鼻窦炎。头皮炎症有时也可成为感染源。抗菌素广泛应用以来,上述病灶的发病率已显著下降,脑脓肿来源于直接蔓延感染者也明显减少。
2.血源性感染病菌来自体内感染灶(肺脓肿、细菌性心内膜炎等)经血流至脑而引起脑脓肿。特别值得注意的是在紫绀性先天性心脏病患者中,脑脓肿的发生率较高,这一情况可能与左右心之间短路的存在以及肺过滤细菌的作用不良有关。
【病理变化】
脑脓肿的部位与数目因感染途径不同而异。一般由局部炎症灶直接蔓延所致的脑脓肿常为单个,其中耳源性(中耳炎、乳突炎)脑脓肿多见于颞叶或小脑;鼻窦(额窦)炎引起的脑脓肿多见于额叶。血源性感染所致者常为多发性,可分布于大脑各部。
脑脓肿的发展规律和形态与全身其它器官的脓肿相似。急性脓肿发展迅速,境界不清,无包膜形成,可向四周扩大,甚至破入蛛网膜下腔或脑室,引起脑室积脓,可迅速致死。慢性脓肿边缘毛细血管和纤维母细胞(源于血管壁)增生明显,并伴有淋巴细胞和巨噬细胞浸润,形成炎性肉芽组织和纤维包膜,境界清楚。脓肿周围脑组织水肿明显,并伴有星形胶质细胞增生(图16-12)。
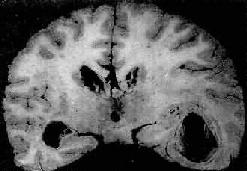
图16-12 慢性脑脓肿
右侧颞叶的脑脓肿与侧脑室相通
【临床病理联系】
脑脓肿的临床表现主要有两个方面:
1.颅内压升高由于脓肿占位性效应及其周围明显的脑水肿所致,临床表现为头痛、呕吐、昏迷、抽搐,小儿前囟饱满。
2.局灶性症状局部脑组织破坏可引起相应的功能障碍,临床表现按病变部位而异,如大脑脓肿可引起半瘫、抽搐,小脑脓肿可引起共济失调等。
【结局】
脓肿小,治疗及时,病灶可被完全吸收而消散。脓肿大,发展迅速,可引起颅内压增高和脑疝形成,后果严重。如脓肿破裂引起脑室炎和脑膜炎,其后果严重,常可致死,如及时进行手术和抗菌治疗,可使死亡率减少到20%以下。
二、病毒性疾病
引起中枢神经系统病毒性疾病的病毒种类繁多,病变可累及软脑膜、脑、脊髓,其中脑脊髓疾病常伴有脑膜反应,故脑脊液中亦可有多少不等的炎性细胞。中枢神经系统病毒性疾病发病前,临床上可有原发感染的前驱症状,如脊髓灰质炎时的非特异性胃肠炎;有的前驱症状不明显,如多灶性白质脑病。
【基本病变】
中枢神经系统病毒性疾病有以下几种基本变化:
(1)炎性细胞浸润:以淋巴细胞,巨噬细胞,浆细胞为主,常环绕血管形成血管套(图16-13)。
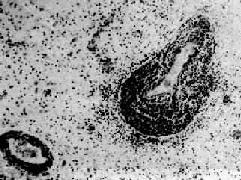
图16-13 血管套
血管周围有炎性细胞环绕
(2)胶质结节形成:这是病毒性脑炎的特征性病变之一。由小胶质细胞和(或)星形胶质细胞增生所致(图16-14)。
图16-14 胶质结节
局部小胶质细胞增生形成胶质结节
(3)包含体形成:多位于神经细胞核中,呈圆形,嗜酸性染色,周围有空晕,核仁被挤向一侧(如单纯疱疹病毒包含体);有的位于胞浆中(如狂犬病Negri小体)。在各种包含体中只有Negri小体具有确诊意义。除神经元外,有些病毒(如进行性多灶性白质脑病的乳多泡病毒)可在少突胶质细胞核中形成包含体。
(4)病变的定位:某种病毒对特定的神经元有一定的亲和性,故病变的定位在中枢神经系统病毒性疾病中是一种较突出的现象,如脊髓灰质炎病毒之与脊髓前角神经元,狂犬病病毒之与海马回神经元,单纯疱疹病毒之与颞叶神经元等。定位的机制不明,可能与细胞表面受体有关。
中枢神经系统病毒感染可借血和(或)脑脊液中抗体、病变组织中病毒颗粒(电镜观察)、特异性病毒抗原(免疫组化法)以及病毒核酸片断(原位分子杂交及多聚酶链反应等)的检出予以确诊。
在中枢神经系统病毒性疾病中最主要的举例如下:
(一)疱疹病毒感染
此病毒为DNA病毒,其中能经起中枢神经系统感染者至少有四种:①单纯疱疹病毒(HSV),②带状疱疹病毒,③EB病毒,④巨细胞病毒,其中以单纯疱疹病毒感染最为多见。
单纯疱疹病毒脑炎是欧美常见的散发性脑炎,可见于新生儿(HSVⅡ型)或见于儿童和青年(HSVⅠ型)。发生在成人中的急性坏死性脑炎病情凶险,死亡率为30%~70%。病变多累及一侧或双侧颞叶或额叶下部,早期病变以坏死性脉管炎和局限性坏死较为突出。进而坏死、出血严重,邻近的脑膜和脑组织炎症反应明显,分别表现为弥漫性淋巴细胞性脑膜炎和血管套形成,胞核中包含体见于神经元及胶质细胞(少突胶质细胞、星形胶质细胞)。存活患者因颞叶损害常引起明显的记忆力丧失,导致严重的痴呆。
(二)肠原病毒感染
最重要的肠原病毒为脊髓灰质炎病毒,柯萨奇(Coxackie)病毒及ECHO病毒,它们都是小型RNA病毒,可引起淋巴细胞性脑膜炎及瘫痪性疾病。后者如脊髓前角灰质炎,多为脊髓灰质炎病毒所致,也可由其他病毒引起。近年来由于积极开展脊髓灰质炎病毒免疫预防,柯萨奇病毒成为此病之常见病因。
脊髓灰质炎(poliomyelitis)又称脊髓前角灰质炎,临床上常伴有肢体瘫痪,故又有小儿麻痹症之称。本病是脊髓灰质炎病毒所引起的散发性或流行性传染病,患者多为儿童。
【病因及传染途径】
脊髓灰质炎病毒有3种亚型,3型间并无交叉免疫。其中Ⅰ型是麻痹性脊髓灰质炎常见病因。此病毒存在于患者的粪便和鼻咽分泌物中,主要经消化道传染,少数也可借飞沫经呼吸道传染。
病毒由消化道侵入机体后,在粘膜上皮内繁殖,然后入血产生暂时性的病毒血症。在机体免疫功能低下时,病毒可侵入中枢神经系统,最后到达靶细胞(运动神经元,特别是脊髓前角运动细胞)。人体感染后,绝大多数表现为隐性感染,少数显性感染病例按其受侵及病变程度之不同,可有3种表现:①轻型:暂时性病毒血症阶段,中枢神经系统未受累,临床上仅有头痛,发热及咽部和肠道症状;②非麻痹型:病毒到达中枢神经系统但仅引起轻微病变,如颈背部肌肉强直,脑脊液中细胞及蛋白质增加(反应性脑脊膜炎),无瘫痪征象;③麻痹型:病毒损害运动性神经元,以脊髓前角运动神经元受损最严重,引起下运动神经元的瘫痪。以上3型中以麻痹型最少见,仅占显性感染的0.1%~1%。
【病理变化】
1.病变的定位分布脊髓运动神经元受累最重,以脊髓颈、腰膨大为甚,其次为大脑前中央回的锥体细胞。除脊髓外,依次而上,病变愈上愈轻:延髓、脑桥、中脑、小脑、下视丘、视丘和苍白球的运动神经元。大脑皮质除前中央回外,很少受累,脊髓后角感觉神经元偶尔也可被累及,但病变轻微。
2.组织变化脊膜显示广泛的充血和炎性细胞浸润,以淋巴细胞和浆细胞为主,有时也可见中性粒细胞;脊髓前角充血、水肿明显,运动神经元有不同程度的变性和坏死(中央性Nissl小体溶解,核浓缩、溶解,鬼影细胞出现,大量神经元脱失)伴淋巴细胞、巨噬细胞、中性粒细胞浸润和小胶质细胞增生。病变晚期噬神经细胞现象突出,并有多量泡沫细胞形成和星形胶质细胞增生,形成胶质瘢痕(图16-15)。
图16-15 脊髓灰质炎
脊髓前角内运动神经细胞变性坏死及消失,胶质细胞增生,并有粒细胞浸润
3.肉眼观脊髓充血明显,脊髓前角充血,病变严重者可显示出血和坏死。晚期,前角萎缩,前根(运动神经根)萎缩、变细。瘫痪的肌肉明显萎缩,肌纤维小,其间为脂肪组织和结缔组织所填充。
【临床病理联系】
由于病变部位和严重程度的不同,临床表现各异。神经元损害须达到一定程度才会出现瘫痪症状,本病以脊髓腰膨大的病变最为严重,瘫痪常发生在下肢;其次为颈膨大,引起上肢瘫痪。脑干的运动神经核受累,可引起颅神经麻痹,如面神经麻痹(Ⅶ,对),软腭瘫痪(Ⅸ,对),声音嘶哑(Ⅹ,对),吞咽困难(Ⅻ,对)等。延髓网状结构受累可引起呼吸、血管运动中枢障碍,导致中枢性呼吸衰竭和循环衰竭而致死。
一般病例发病后1~2周即进入临床恢复期,瘫痪肢体开始有不同程度的恢复。未能完全恢复者,患者肌肉逐渐萎缩,成为后遗症。
(三)虫媒病毒感染
目前已知的虫媒病毒有数百种之多,均为RNA病毒,其中能引起较严重疾病者有十余种。在我国常见的是由蚊传播的乙型脑炎和蜱传播的森林脑炎。
流行性乙型脑炎(epidemic encephalitis B)是乙型脑炎病毒感染所致的急性传染病,多在夏季流行,儿童发病率明显高于成人,尤以10岁以下儿童为多,约占乙型脑炎的50%~70%。乙型脑炎之称在于与冬季发生的昏睡性甲型脑炎相区别,后者自1925年后已无流行,仅有散发病例出现。
【病因及传染途径】
乙型脑炎病毒为RNA病毒,其传播媒介为蚊(在我国主要为三节吻库蚊)和长期贮存宿主。在自然界,其循环规律为:动物-蚊-动物,在牛、马、猪等家畜中隐性感染率甚高,一般仅出现病毒血症,成为人类疾病的传染源和贮存宿主。带病毒的蚊叮人吸血时,病毒可侵入人体,先在局部血管内皮细胞及全身单核吞噬细胞系统中繁殖,然后入血引起短暂性病毒血症。病毒能否进入中枢神经系统,取决于机体免疫反应和血脑屏障碍功能状态。凡免疫能力强,血脑屏障碍功能正常者,病毒不能进入脑组织致病,故成为隐性感染,多见于成人。在免疫功能低下,血脑屏障功能不健全者,病毒可侵入中枢神经系统而致病,由于受染细胞表面有膜抗原存在,从而激发体液免疫与细胞免疫,导致损伤和病变的发生。
【病变】
本病病变广泛累及整个中枢神经系统灰质,但以大脑皮质及基底核、视丘最为严重,小脑皮质、延髓及脑桥次之,脊髓病变最轻,常仅限于颈段脊髓。
肉眼观,脑膜充血,脑水肿明显,脑回宽,脑沟狭;切面上在皮质:深层、基底核、视丘等部位可见粟粒大小的软化灶,其境界清楚,弥漫分布或聚集成群。
镜下,可出现以下病变:
1)血管变化和炎症反应血管高度扩张充血,可发生明显的淤滞,血管周围间隙增宽,脑组织水肿,有时可见环状出血。灶性炎性细胞浸润多以变性和坏死的神经元为中心,或围绕血管周围间隙形成血管套(图16-13)。浸润的炎性细胞以淋巴细胞、单核细胞和浆细胞为主,仅在早期有为数不多的中性粒细胞。
2)神经细胞变性、坏死病毒在神经细胞内增殖,导致细胞的损伤,表现为细胞肿胀,Nissl小体消失,胞浆内空泡形成,核偏位等。病变严重者神经细胞可发生核浓缩、溶解、消失,为增生的少突胶质细胞所环绕,出现卫星现象。此外,噬神经细胞现象也很常见。
3)软化灶形成灶性神经组织的坏死、液化,形成镂空筛网状软化灶,对本病的诊断具有一定的特征性。病灶呈圆形或卵圆形,边界清楚(图16-16),分布广泛,除大脑(顶叶、额叶、海马回)皮质灰、白质交界处外,丘脑、中脑等处也颇常见。关于软化灶发生的机制至今尚未能肯定,除病毒或免疫反应对神经组织可能造成的损害外,病灶的局灶性分布提示,局部循环障碍(淤滞或小血管中透明血栓形成)可能也是造成软化灶的一个因素。
图16-16 流行性乙型脑炎
脑组织内有多发性镂空软化灶
4)胶质细胞增生小胶质细胞增生明显,形成小胶质细胞结节(图16-14),后者多位于小血管或坏死的神经细胞附近。少突胶质细胞的增生也很明显。星形胶质细胞增生和胶质瘢痕形成,在亚急性或慢性病例中较为多见。
【临床病理联系】
本病病变分布广泛,神经细胞广泛受累,患者常出现嗜睡、昏迷以及颅神经核受损所致的颅神经麻痹症状。由于脑内血管扩张充血,血流淤滞、血管内皮细胞受损,致血管通透性增高而引起脑水肿和颅内压升高,病人常出现头痛、呕吐。严重的颅内压增高可引起脑疝,其中小脑扁桃体疝可致延髓呼吸中枢受压而致死。由于脑膜有不同程度的反应性炎症,临床上有脑膜刺激症状和脑脊液中细胞数增多的现象。
本病患者经过治疗,大多数在急性期后可痊愈,脑部病变逐渐消失。病变较重者,可出现痴呆、语言障碍、肢体瘫痪及颅神经麻痹引起的吞咽困难、中枢神经性面瘫、眼球运动障碍等,这此表现经数月之后多能恢复正常。少数病例病变不能完全恢复而留下后遗症。
(四)狂犬病
狂犬病(rabies)是狂犬病毒引起的传染病。狂犬病的流行于犬、猫等(狼、蝙蝠等亦可感染),成为狂犬病毒的贮存宿主。人被病犬(或病猫等)咬伤,唾液中的病毒经伤口侵入人体,沿周围神经(主要是感觉神经)至背根节经脊髓入脑而致病。
狂犬病的潜伏期随伤口的部位而异,如面部咬伤者潜伏期较下肢被咬伤者要短得多。本病临床表现为创口疼痛、头痛、发热、不安、怕风,饮水时反射性咽喉痉挛,故有恐水病之称。后期可出现昏迷、呼吸衰竭。
【病理变化】
脑和脊髓充血明显,病变一般在神经根节、脑干下端、下丘脑等部位最为显著。镜下,可见弥漫性急性脑脊髓炎变化,有不同程度的神经元损害,从变性到坏死及神经细胞被吞噬;血管周围有淋巴细胞、浆细胞浸润,形成血管套。神经细胞内出现特征性的Negri小体具有病理诊断意义。该小体是神经细胞浆内的包含体,呈圆形或椭圆形,边界清楚,体积大,呈嗜酸性着色,多见于海马锥体细胞、小脑Purkinje细胞和大脑锥体细胞。每一神经细胞胞浆中小体的数目从一个到数个不等(图16-17)。炎症病变严重的部位,Negri小体数目往往不多。
图16-17 狂犬病
神经细胞胞浆中Negri小体
Negri小体含有病毒核壳体(核酸与蛋白质外膜组成),可用免疫组化加以显示,但电镜下看不到病毒颗粒。由于中枢神经系统中的病毒可沿周围神经离心性播散而侵入其他组织,故用免疫组化染色法可从皮肤活检或角膜细胞中检出Negri小体。当患者疑为疯狗咬伤时,应保留该狗数天,处死后检查其脑部,寻找Negri小体以帮助确诊。
(五)人类免疫缺陷病毒感染
人类免疫缺陷病毒(human immunodeficiency virus,HIV)除选择性地破坏CD4+T细胞外,中枢神经系统也是HIV的靶器官,约有70%AIDS患者出现中枢神经系统症状。与AIDS相关的神经系统病变有下列数种:
1.病毒性脑膜炎 本病变出现在HIV感染的早期,可无临床症状,或症状可自行缓解,表现为蛛网膜淋巴细胞和巨噬细胞浸润,脑脊液中单个核细胞数目增加,并出现HIV特异抗体。目前认为中枢神经系统的HIV感染是由受病毒感染的血源性巨噬细胞带入所致,因此感染早期的病毒性脑膜炎为HIV日后引起中枢神经系统病变创造了条件。
2.周围神经病变病变主要表现为脱髓鞘,严重时可伴有轴索的破坏,导致感觉和(或)运动障碍。临床表现可为自限性Guillian-Barre综合征,颅神经或周围神经炎;如累及背根神经节,可引起共济失调。约有90%处于潜伏期的HIV感染者可以周围神经损害为其唯一表现。
3.空泡性脊髓病(vacuolar myelopathy)病变主要累及脊髓后索和侧索。可见局部髓鞘肿胀,出现空泡、脱髓鞘伴巨噬细胞浸润和反应性星形胶质细胞增生。偶见多核巨细胞。患者可呈现进行性下肢瘫痪,感觉性共济失调和大小便失禁。约有20%~30%的AIDS尸检病例可检出此病变。
4.亚急性脑炎 亚急性脑炎是AIDS的主要中枢神经系统病变,约有30%的AIDS尸检病例可检出此病变。病变主要累及大脑和小脑的白质,以及深部皮质。在程度不等的脱髓鞘病灶中可见巨噬细胞浸润,多核巨细胞形成(图16-18),在某些多核巨细胞胞浆中可检出HIV病毒颗粒或病毒蛋白(如P24)。淋巴细胞浸润程度较轻,主要在血管周围,且多为CD8+T细胞。此外还可有反应性星形胶质细胞增生,神经元也有一定程度的缺失。临床上可出现进行性精神和行为异常,精神淡漠,共济失调,震颤,终致出现AIDS痴呆综合征。然而痴呆的确切发病机制尚待阐明。
图16-18 AIDS亚急性脑炎
示病灶内形成的不典型多核巨细胞
中枢神经系统的常见机会感染为:巨细胞病毒、带状疱疹病毒、单纯疱疹病毒脑炎,进行性多灶性白质脑病,隐球菌和弓形虫感染等。
(六)慢病毒感染
大多数慢病毒感染疾病均发生在中枢神经系统,其临床特点为潜伏期长(数月至数十年不等),起病缓慢,呈进行性、致死性发展。这类疾病有十余种,发生在人类者有4种。
1.亚急性硬化性全脑炎本病是中枢神经系统持续性麻疹病毒感染所致,患者多为儿童和青少年(4~20岁),有麻疹病史,临床表现为精神和运动障碍、肌阵挛、抽搐,最后出现痴呆和去皮质强直。
病理变化:肉眼观,脑质地较坚硬,部分区域呈颗粒状。镜下,神经细胞被吞噬,血管周围单核细胞浸润;星形胶质细胞增生,神经元和少突胶质细胞核内包含体形成;电镜下可见包含体内有类似麻疹病毒的颗粒。
脑组织中持续存在病毒颗粒可能与宿主T细胞抑制有关,也可能由于病毒本身M蛋白的缺乏,致病毒的装配、释放发生障碍,使感染持续存在。M蛋白缺乏的原因至今尚不明了。同于麻疹疫苗的广泛使用,因缺陷麻疹病毒引起的本病已极为少见。
2.进行性多灶性白质脑病本病是一种乳多泡病毒所致之机会性中枢神经系统感染,见于晚期造血系统恶性肿瘤,应用免疫抑制剂、免疫缺陷病(AIDS)和慢性消耗性疾病(结核病、结节病、类风湿性关节炎)等患者。临床上,本病往往出现在上述各类疾病后数月或数年,患者多为中年人,但也可见于儿童。
本病病变的特点是脑白质中不对称分布的多发性不规则形灰色透明凹陷病灶,质地软,严重者病灶可融合,甚至呈囊性变。镜下可见大小不一的脱髓鞘病灶,其中有多少不一的泡沫细胞。特异性变化包括:①少突胶质细胞的异常是本病有诊断意义的变化,表现为核大而深染,其中有紫色或嗜伊红色包含体。电镜下可见核内有多量乳多泡病毒样颗粒,②星形胶质细胞大而奇异,多核浓染。与本病有关之乳多泡病毒为JC、BK和SV40病毒。由于约65%正常人血清中可检出其特异性抗体,本病的发生究竟是陈旧感染复燃,还是易感宿主被感染尚未阐明。
3.海绵状脑病本病为一组疾病,其共同特点是脑灰质疏松呈海绵状,其中亚急性海绵状脑病和Kuru病可见于人类和灵长类。此类疾病的病原体为小分子量(60000)的糖蛋白称为Prion(Pr)。Pr是正常神经元的膜蛋白,本身并不致病。如其结构发生一个氨基酸的变异,形成Pr5C则不能被蛋白酶完全降解,形成Pr27-30。后者形成类淀粉蓄积于神经元胞体内,造成神经元死亡。Pr27-30感染(接种)于另一个体可起模板作用,进行大量复制,造成疾病蔓延。Kuru病仅发生在大洋洲巴布亚新几内亚,由于食人尸脑而传播。革除陋习后此病几乎已绝迹。
亚急性海绵状脑病 又名Creutsfield-Jacob disease(CJD)病,是少见的世界性散发疾病,表现为进行性痴呆。预后差,无有效治疗。在人类中其传播途径尚不清楚,经角膜移植所致之医源性感染的病例已有报道。本病最突出的病理变化是大脑各叶(以额叶、颞叶为甚)均有极明显的神经细胞脱失,伴星形细胞高度增生,而无炎症反应。脑皮质疏松呈海绵状,白质大多正常。有时纹状体、丘脑、小脑表层等处亦可发生类似变化。电镜下可见海棉空隙乃神经元和胶质细胞突起中的空泡形成。
第三节 缺氧与脑血管病变
脑血管疾病的发病率和死亡率在国内、外均名列前茅。脑重量仅为体重的2%,但其耗氧量则占全身耗氧量的20%,其所需血供占心输出量的15%。加之脑组织不能储存能量,也不能进行糖的无氧酵解,因此其对氧和血供的要求特别高。缺血缺氧4分钟即可造成神经元的死亡。尽管机体存在一系列的代偿调节机制(如脑底动脉环的存在可使局部缺血区域得到一定程度的供血补偿;缺血缺氧时脑血管扩张,全身其它器官血管收缩以进行血液重新分配等),但这种调节机制仍有一定的限度,一旦超过此极限,即可造成神经元损伤。
一、缺血性脑病
缺血性脑病(ischemic encephalopathy)是指由于低血压、心脏骤停、失血、低血糖、窒息等原因引起的脑损伤。
【影响病变的因素】
脑的不同部位和不同的细胞对缺氧的敏感性不尽相同。不脑较脑干各级中枢更为敏感,大脑皮质较白质敏感。各类细胞对缺氧敏感性由高至低依次为:神经元、星形胶质细胞、少突胶质细胞、内皮细胞。神经元中以皮质第Ⅲ、Ⅴ、Ⅵ层细胞,海马锥体细胞和小脑蒲肯野细胞最为敏感,在缺血(氧)时首先受累。
脑损伤程度取决于缺血(氧)的程度和持续时间以及患者的存活时间。轻度损伤往往无明显病变,重度损伤患者仅存活数小时者尸检时也可无明显病变。只有中度损伤,存活时间在12小时以上者才出现典型病变。
此外,损伤的部位还和局部的血管分布和血管的状态有关。在发生缺血(氧)时,动脉血管的远心端供血区域最易发生灌流不足。大脑分别由来自颈内动脉的大脑前动脉、大脑中动脉和来自椎动脉的大脑后动脉供血。其中大脑前动脉供应大脑半球的内侧面和大脑凸面的额叶、顶叶近矢状缝宽约1~1.5cm的区域。大脑中动脉则供应基底核、纹状体、大脑凸面的大部区域。而大脑后动脉则供应颞叶的底部和枕叶。这样在3支血管的供应区之间存在一个C形分布的血供边缘带,该带位于大脑凸面,与矢状缝相平行,且旁开矢状缝1~1.5cm(图16-19)。一旦发生缺血性脑病,该区域则最易受累。然而并非每例缺血性脑病病灶都呈C型,病灶的形状还受局部血管管径的影响,如果某支血管管径相对较小,或局部动脉粥样硬化,则其供血区较易受累。
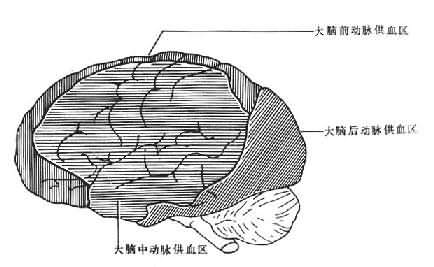
图16-19大脑前、中、后动脉供血区之间的C形边缘带示意图
【病理变化】
脑缺血的组织学变化在缺血12小时以后才较明显:神经元出现中央性Nissl小体溶解和坏死(红色神经元);髓鞘和轴突崩解;星形胶质细胞肿胀。1~2天出现脑水肿,中性粒细胞和巨噬细胞浸润,并开始出现泡沫细胞。第4天星形胶质细胞明显增生,出现修复反应。大约30天左右形成蜂窝状胶质瘢痕。
缺血性脑病的常见类型:①层状坏死。大脑皮质第3、5、6层神经元坏死、脱失、胶质化,引起皮质神经细胞层的中断。②海马硬化。海马锥体细胞损伤、脱失、胶质化。③边缘带梗死(图16-20)。梗死的范围与血压下降的程度和持续的时间有关,如血压持续下降,则梗死区自远心端向次远心端扩大,称为向心性发展(cardiopetal development),即C形梗死区向其两侧扩大,并自大脑顶部向颅底发展。大脑缺血性脑病边缘带梗死的极端情况是全大脑的梗死,但脑干的各核团由于对缺血(氧)的敏感性较低仍可存活。患者靠呼吸器以维持生命,但意识丧失,成为植物人。如何处置这样的病人则成为目前医学伦理学和医疗实践的难题。一旦这种病人死亡,其大脑乃成为由脑膜包裹,秽暗无结构的坏死组织,称为呼吸器脑。
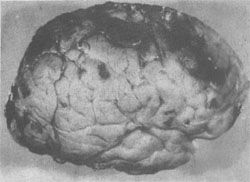
图16-20 大脑缺血性脑病
大脑前、中、后动脉血供边缘带出血性梗死灶呈C形
综上所述,缺血性脑病的临床表现因缺血的严重程度和持续时间而异,轻者仅发现一过性精神错乱,或出现上肢肩带肌力和感觉减退,重者则可昏迷死亡。
二、脑梗死
脑梗死是由于血管阻塞引起局部血供中断所致。大动脉,如颈内动脉、椎动脉之间存在脑底动脉环,故其中一支阻塞时一般不致引起梗死。中等大动脉,如大脑前动脉、大脑中动脉等,其终末支之间仅有部分吻合,血管管腔阻塞可导致梗死,但梗死区小于该血管供应区。小动脉,如豆纹动脉、皮质穿支则少有吻合支,一旦发生阻塞,则梗死的范围和血管供应区基本一致。
引起脑梗死的血管阻塞,可以是血栓性阻塞,也可以是栓塞性阻塞。
(一)血栓性阻塞
发生在动脉粥样硬化的基础上,粥样硬化好发于颈内动脉与大脑前动脉、中动脉分支处,及后交通动脉、基底动脉等处。粥样斑块本身、斑块内出血、附壁血栓均可阻塞血管。这种阻塞发展较慢。在发生血管阻塞以前患者可有一过性的局部的神经系统症状或体征,称为一过性脑缺血症(transient ischemic attacks,TIAs)。血栓性阻塞所致脑梗死其症状常在数小时或数天内不断发展,表现为偏瘫、神态不清、失语等。
(二)栓塞性阻塞
栓子可来源于全身各处,但以心源性栓子居多。病变常累及大脑中动脉供应区。其发生往往比较突然,以致临床表现急骤,预后也较差。
【病变】
脑梗死有贫血性和出血性之分。由于局部动脉血供中断引起的梗死一般为贫血性。但如果其后梗死区血供又有部分恢复(如栓子碎裂并随再通血流运行)则再灌流的血液可经遭缺氧损害的血管壁大量外溢,使贫血性梗死转变成出血性。大静脉(如矢状窦)血栓形成先引起组织严重淤血,继之发展为淤血性梗死,亦属出血性梗死。
脑梗死的肉眼观变化要在数小时后才能辨认。梗死区灰质暗淡,灰质白质界线不清。2~3天后局部水肿,夹杂有出血点。一周后坏死组织软化,最后液化形成蜂窝状囊腔。组织学变化与缺血性脑病基本一致。值得指出的是,由于脑膜和皮质之间有吻合支存在,故梗死灶内皮质浅层的分子层结构常保存完好,这是脑梗死和脑挫伤的形态学鉴别要点。
腔隙状坏死(lacunae)是直径小于1.5cm的囊型病灶,常呈多发性。可见于基底核、内囊、丘脑,脑桥基底部与大脑白质。引起腔隙状坏死的原因,可以是在高血压基础上引起的小出血,也可以是深部细动脉阻塞(栓塞或高血压性血管玻璃样变)引起的梗死。除非发生在特殊的功能区,腔隙状坏死可无临床表现。
三、脑出血
颅内出血包括硬脑膜外出血、硬脑膜下出血(详本章创伤节)和脑出血。后者可分成脑内出血、蛛网膜下出血和混合性出血。
(一)脑内出血
高血压病是脑内出血(intracerebral hemorrhage)的最常见原因,其发生机制详见高血压病。此外此类出血也可见于血液病、血管瘤破裂等。
大块型脑出血常急骤起病,患者突感剧烈头痛,随即频繁呕吐、意识模糊,进而昏迷,神经系统体征依出血的部位和出血范围而定。基底核外侧型出血常引起对侧肢体偏瘫,内侧型出血易破入侧脑室和丘脑,脑脊液常为血性,预后极差。脑桥出血以两侧瞳孔极度缩小呈针尖样为特征。小脑出血则出现出血侧后枕部剧痛及频繁呕吐。脑内出血的直接死亡原因多为并发脑室内出血或严重的脑疝。
(二)蛛网膜下腔出血
自发性蛛网膜下腔出血(subarachnoid hemorrhage)约占脑血管意外的10%~15%。临床表现为突发剧烈头痛,脑膜刺激症状和血性脑脊液,其常见的原因为先天性球性动脉瘤,好发于基底动脉环的前半部,并常呈多发性(图16-21),因此有些患者可多次出现蛛网膜下腔出血。先天性球性动脉瘤常见于动脉分支处,由于该处平滑肌或弹力纤维的缺如,在动脉压的作用下膨大形成动脉瘤。动脉瘤一旦破裂,则可引起整个蛛网膜下腔积血。大量出血可导致患者死亡,机化的蛛网膜下腔出血则可造成脑积水。
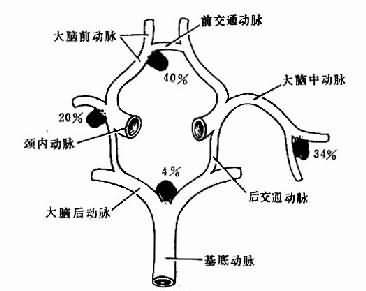
图16-21 基底动脉环各处先天性球性动脉瘤的发生率
(三)混合性出血
常由动静脉畸形(arteriovenous malformations,AVMs)引起,AVMs是指走向扭曲,管壁结构异常,介于动脉和静脉之间的一类血管,其管腔大小不一,可以成簇成堆出现。约90%AVMs分布于大脑半球浅表层,因此其破裂常导致脑内和蛛网膜下腔的混合性出血。患者除出现脑出血和蛛网膜下腔出血的表现外,常可有癫痫史。
第四节 创伤
颅脑损伤多见于男性青壮年,是严重创伤的主要死因。颅脑损伤包括颅骨骨折,脑膜损伤和脑实质损伤。
一、脑膜损伤
脑膜损伤主要引起出血。
(一)硬脑膜外出血
硬脑膜外出血(epidural hemorrhage,EDH)此种出血以颞部较多见。创伤局部或颞骨骨折导致脑膜中动脉撕裂出血。血肿常引起颅骨与局部硬脑膜分离,压迫局部脑组织,形成平整而边界不清的压迹。典型的临床表现是患者在因创伤所致的短时意识丧失后,有6~8小时的清醒期,随着血肿的形成和发展,患者再次陷入进行性昏迷状态。患者常因脑疝、呼吸衰竭死亡,因此应及时确诊并进行手术处理。
(二)硬脑膜下出血
硬脑膜下出血(subdural hemorrhage,SDH)多因桥静脉(连接脑皮质和上矢状窦)撕裂所致,因此出血位置多在大脑背侧部,在硬脑膜和蛛网膜之间。血肿大小与机体的凝血机能和出血的次数等有关。局部大脑受压,由于血肿直接压迫脑组织,致使压迹凹陷呈不规则状,轮廓分明(图16-22)。
图16-22 硬脑膜外血肿与硬脑膜下血肿示意图
急性硬脑膜下血肿可伴有脑挫伤或撕裂。临床症状出现较缓慢,有不同程度的意识障碍。其后果取决于出血程度及局部脑损伤等因素。
慢性硬脑膜下血肿常发生在轻微脑损伤后,多见于具有脑萎缩之老年人。起病缓慢,往往到血肿发展至相当体积后才出现症状。可表现为精神错乱,注意力不集中,偶可出现癫痫和缓慢进行的昏迷。血肿表面有起源于硬脑膜的肉芽组织包围。轻微损伤又可使其中的毛细血管破裂导致少量出血,这种出血、机化的过程可反复进行,使肉芽组织机化呈层状增厚,并使血肿进行性增大。未经治疗者多死于因颅压升高所致的脑损害。
(三)蛛网膜下腔出血
蛛网膜下腔出血(subarachnoid hemorrhage,SAH)可伴发于脑挫伤,也可单独存在。通常出血范围有限,少数可为广泛、弥漫的出血。广泛弥漫的蛛网膜下腔出血、机化,可引起脑积水。
二、脑实质损伤
脑实质损伤有下列几种。
(一)脑震荡
脑震荡(concussion)是头部创伤后暂时性意识丧失。其发生可能与中脑旋转所致网状系统一过性功能障碍有关。一般无明显的结构变化。必须指出的是,临床医生单凭症状作脑震荡的诊断须相当慎重。不少有脑震荡脑史的患者,在其尸检时可发现程度不同的脑挫伤。
(二)脑挫伤
脑挫伤(contussion)和撕裂是最常见的局限性脑损伤。脑损伤发生在直接受外力冲击之处称为冲击伤(coup lesion),发生在其对侧者称为对冲伤(contra coup)。后者的发生和脑在受损过程中的旋转和位移有关。对冲伤易发生在额叶、颞叶,而枕叶甚少见。此与颅底不规则骨性粗糙面有关。
脑挫伤多累及脑回之冠,脑沟深部大多完好。病变呈楔形,底朝表面,尖端位于深层。局部软脑膜和皮层全层坏死(皮质分子层坏死是与脑梗死相区别的特征),并伴皮层血管撕裂出血。挫伤灶最后由增生的星形胶质细胞和由软脑膜纤维母细胞增生形成的纤维胶质疤痕加以修复,病灶和硬脑膜粘连。
(三)脑撕裂
脑撕裂(laceration)乃由头部重度钝器伤造成,除脑皮质外病灶还累及深部脑组织。
(四)弥漫性轴突损伤
弥漫性轴突损伤(diffuse axonal injury)患者在颅脑损伤后即出现深昏迷和植物状态。肉眼观脑无明显病变。镜下,轴突广泛肿胀,以大脑白质、胼胝体和脑干上部最为显著。继之出现髓鞘变性,灶性出血坏死和小胶质细胞增生。本病的发病机制可能与加速或减速过程中对脑造成的剪切力(shearing force)损伤轴突有关。多见于汽车车祸。约有20%患者经治疗可恢复正常意识。
(五)脑出血
损伤性脑出血常伴发于脑挫伤、撕裂伤和急性轴突损伤。一般为点状或灶性出血。如大血管撕裂则可导致大出血或血肿形成。
第五节 肿瘤
原发性中枢神经系统肿瘤发病率约2~5/10万,占肿瘤尸检率的13%,其中85%为颅内肿瘤,15%为椎管内肿瘤。
原发性颅内肿瘤以胶质瘤最为多见,约占40%,其次为脑膜瘤(13%~19%),听神经瘤(神经鞘瘤)(8%~9%)。胶质瘤中恶性星形胶质细胞瘤(胶质母细胞瘤)占50%~60%,良性星形胶质细胞瘤约占25%~30%。
儿童的颅内肿瘤发病率甚高,仅次于白血病而占小儿恶性肿瘤的第2位。除胶质瘤外,髓母细胞瘤最常见,而脑膜瘤和神经鞘瘤则甚罕见。约70%的肿瘤位于天幕下(成人70%发生在天幕上),脊髓肿瘤甚罕见。常见神经系统肿瘤见表16-1。
表16-1 神经系统常见肿瘤
| 中枢神经系统肿瘤 |
| 胶质瘤 |
| 星形胶质细胞瘤 |
| 少突胶质细胞瘤 |
| 室管膜细胞瘤 |
| 脉络丛乳头状瘤 |
| 原始神经上皮源性肿瘤 |
| 髓母细胞瘤 |
| 脑膜瘤 |
| 松果体肿瘤 |
| 垂体肿瘤 |
| 周围神经肿瘤 |
| 神经鞘膜肿瘤 |
| 神经鞘瘤 |
| 神经纤维瘤 |
| 神经细胞源性肿瘤 |
| 神经母细胞瘤 |
| 节细胞神经瘤 |
| 转移性肿瘤 |
颅内肿瘤可引起以下症状:①肿瘤压迫或破坏周围脑组织所引起的局部神经症状,如癫痫、瘫痪、视野缺损等。②颅内占位病变引起颅内压增高的症状,表现为头痛、呕吐、视神经乳头水肿等。
一、中枢神经肿瘤
(一)胶质瘤
胶质瘤(glioma)具有特异的不同于其它部位肿瘤的生物学特性:
(1)良恶性的相对性:无论高度分化或低度分化的胶质瘤均呈浸润性生长,更无包膜形成。生长迅速、间变程度高的肿瘤,与周围组织截然不同,故边界往往较清楚。
第三脑室分化良好的幼年型星形胶质细胞瘤,由于位于手术禁区,无法进行切除。因此预后不佳。
(2)局部浸润:胶质瘤的浸润性生长主要累及血管周围间隙、软脑膜、室管膜、神经纤维束间。
(3)转移:①脑脊液转移是颅内肿瘤常见的转移方式,相当于颅外恶性肿瘤细胞的淋巴道浸润和转移,特别是位于脑室旁、脑池旁的肿瘤发生这种转移的机会更多。②颅外转移极少见,其中80%以上均有颅脑外科手术史。
1.星形胶质细胞瘤(astrocytoma) 本瘤约占颅内肿瘤的30%,占神经胶质瘤的70%以上。男性较多见。最近研究表明该肿瘤中原癌基因sis有过度表达,erb-B1则有放大。
肉眼观,肿瘤为数厘米的结节至巨大块状。分化较好的肿瘤,境界不清;而分化程度较低的肿瘤则境界分明。瘤体灰白色。质地视肿瘤内胶质纤维多少而异,或硬、或软、或呈胶冻状外观,并可形成大小不等的囊腔。由于肿瘤的生长、占位和邻近脑细胞的肿胀,脑的原有结构因受挤压而扭曲变形(图16-23)。
图16-23 星形胶质细胞瘤
左大脑半球肿胀,肿瘤边界不清,部分呈胶冻状
镜下,肿瘤细胞形态多样,可相似于纤维型星形胶质细胞、原浆型星形胶质细胞和肥胖星形胶质细胞,故分别称为纤维型、原浆型和肥胖型星形胶质细胞瘤。前二者为良性肿瘤,后者性质介于良恶性之间。如肿瘤细胞出现间变,细胞密度增大,异型性明显,核深染,出现核分裂像,毛细血管内皮细胞增生,则为间变性星形胶质细胞瘤,为恶性肿瘤。
高度恶性的星形胶质细胞瘤称为多形性胶质母细胞瘤(glioblastoma multiforme),多见于成人。肿瘤好发于额叶、颞叶白质,浸润范围广,常可穿过胼胝体到对侧,呈蝴蝶状生长(图16-24)。瘤体因常有出血坏死而呈红褐色。镜下,细胞密集,异型性明显,可见怪异的单核或多核瘤巨细胞。出血坏死明显,是其区别于间变性星形胶质细胞瘤的特征。毛细血管明显增生,内皮细胞增生、肿大,可导致管腔闭塞和血栓形成。肿瘤发展迅速,预后极差,患者多在2年内死亡。
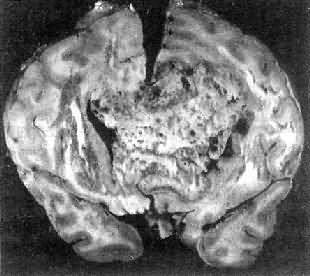
图16-24 多形性胶质母细胞瘤
在两半球内肿瘤呈蝴蝶状,边界不清,切面见有出血、坏死及液化
发生于儿童、青少年的毛发细胞型星形胶质细胞瘤,生长极为缓慢。有报道称患者在不完全切除肿瘤后有带瘤存活达40年者。该瘤常位于小脑、第四脑室底部、第三脑室、丘脑和视神经。其形态特点是由双极性的肿瘤细胞两端发出纤细的毛发状突起。即使有毛细血管增生,本瘤的预后仍相对较好。
应该指出,同一肿瘤的不同区域,瘤细胞可有不同的形态特征,且分化程度也不尽相同,因此肿瘤的分型仅具有相对的意义。
星形胶质细胞瘤的细胞骨架含有胶质纤维酸性蛋白(GFAP),免疫组织化学染色呈阳性反应,是该肿瘤的特异标志。
2.少突胶质细胞瘤(oligodendroglioma) 约占颅内胶质瘤的5%,主要见于30~40岁的成人,男女发病的机会相等。本瘤绝大部分位于大脑半球皮质的浅层,尤以左额叶为多见。
肉眼观,肿瘤呈灰红色边界清楚的球形肿块,位于白质和邻近的皮质,并可累及软脑膜。囊性变、出血和钙化颇为常见,其中钙化灶对X线诊断有一定帮助。
镜下,瘤细胞大小均匀,形态单一,弥漫排列,胞核居中着色深,胞浆空,环绕胞核形成空晕。间质富有血管,有不同程度的内皮细胞增生。约有20%病例可出现瘤细胞钙化,其范围大小不一,其中7%为镜下钙化,有时钙化灶较大,可在X线片上显示出来。如肿瘤组织中混有星形胶质细胞瘤成分达到50%,则称混合性少突星形胶质细胞瘤。
本瘤生长缓慢,病程可长达10~30年,临床表现多为癫痫和局部性瘫痪。少数生长迅速,酷似多形性胶质母细胞瘤,预后不佳。
3.室管膜(细胞)瘤(ependymoma)起源于室管膜细胞,患者多为儿童和青少年。本瘤占颅内胶质瘤的5%~6%,多见于第四脑室,其次为侧脑室、第三脑室和导水管。脊髓病变多发生于腰胝及马尾部。
肉眼观,颅内室管膜瘤呈膨胀性生长,边界清楚,呈球形、分叶状或乳头状,肿瘤多在脑室内生长。切面灰白色,呈均匀或颗粒状,可发生灶性出血甚至坏死或囊性变,有时也可发现点状钙化。
镜下,瘤细胞大小形态一致,呈梭形或胡萝卜形,胞核圆或椭圆,染色质呈细颗粒状,核膜清楚,有核仁。瘤细胞胞浆丰富,突起明显。瘤细胞的排列有二种特征,一是环绕空腔排列成腺管状,形态上与室管膜腔相似,称为菊形团形成,另一是环绕血管形成假菊形团结构,瘤细胞有细长的胞浆突起与血管壁相连(图16-25)。细胞中有神经胶质纤维,以PTAH染色在个别细胞的腔面或胞核旁可见纤毛体,后者与纤毛运动有关,是室管膜细胞的特征性结构。此外,有时还可形成乳头状结构。发生在脊髓圆锥和终丝的肿瘤,乳头状结构轴心中的结缔组织往往富含粘液。
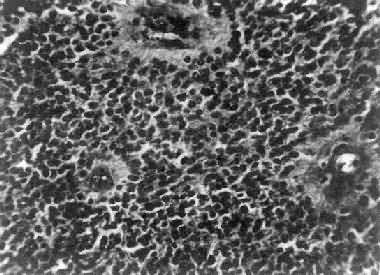
图16-25 室管膜瘤
瘤细胞为圆形或卵圆形,核染色质丰富,胞浆少,可见有细长的胞浆突起与血管相连,呈放射关,形成假菊形团
(二)髓母细胞瘤
髓母细胞瘤(medulloblastoma)好发于儿童,仅次于星形胶质细胞瘤而占第2位,其发生率占儿童颅内肿瘤的25%,发病年龄75%为15岁以下,偶见于成人,男性较女性为多(2~3:1)。
本瘤来源于小脑蚓部的原始神经上皮细胞或小脑皮质的胚胎性外颗粒层细胞,故本瘤主要见于小脑,在儿童多发生于小脑蚓部,在成人则多见于小脑半球。
肉眼观,瘤组织呈鱼肉状,色灰红。镜下,肿瘤由圆形、椭圆形或胡萝卜形细胞构成,胞核着色深,胞浆少而边界不清楚,有多少不等的核分裂像。细胞密集,间质中有纤细的纤维,血管不多。瘤细胞环绕一个嗜银性纤细的神经纤维中心作放射状排列形成典型的菊形团(图16-26),这对髓母细胞瘤的病理诊断有一定的意义。瘤细胞具有向神经元及神经胶质双向分化的潜能,既能向神经母细胞、节神经细胞分化,也能向胶质母细胞、星形胶质细胞分化。如瘤细胞侵入软脑膜,可在蛛网膜下腔脑脊液中广泛播散转移。
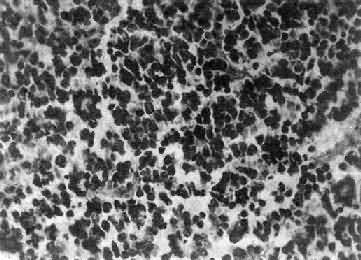
图16-26 髓母细胞瘤
瘤细胞较小,着色深,密集排列,有菊形团形成
髓母细胞瘤恶性程度高,预后差。
(三)脑膜瘤
脑膜瘤(meningioma)可来源于脑膜的各组成成分如蛛网膜细胞,纤维母细胞或血管,其中多数来源于蛛网膜颗粒中的蛛网膜细胞。本瘤大多生长缓慢,良性类型可完全无症状,在70岁以上老人的尸检中,发现无症状的脑膜瘤不在少数,无症状脑膜瘤占颅内肿瘤的14%。脑膜瘤患者多为40~50岁中年人,女性较男性多。
肿瘤的好发部位与蛛网膜颗粒所在部位相同,常见于上矢状窦旁大脑镰两侧,蝶骨嵴,嗅沟,小脑脑桥角;在脊髓则以胸段为多见,一般颅内脑膜瘤较脊髓脑膜瘤多2倍。
肉眼观,肿瘤呈球形,分叶状或不规则形,质实或硬,边界清楚,周围脑组织受压成凹陷切迹(图16-27)。少数肿瘤呈斑块状覆盖较广泛区域,甚至整个脑半球,称为斑块型脑膜瘤。肿瘤质地硬,切面灰白色,呈颗粒状、条索旋涡状,有的质地似砂砾样,乃由于有多量砂粒体存在。
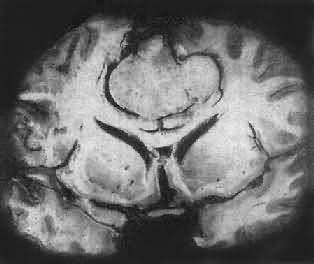
图16-27 脑膜瘤
于大脑两半球间有一近似球形肿瘤,边界清楚,周围脑组织受压萎缩
由于脑膜瘤的组织来源复杂,其组织学图像也可呈现下列基本类型:
(1)脑膜细胞型(融合细胞型):瘤细胞胞浆丰富,边界不清楚,胞核椭圆形,细胞排列呈分叶状或旋涡状,为纤维间质条索所分隔。
(2)纤维细胞型:瘤细胞呈长梭形,排列成致密的交织束状结构,其间有网状纤维及胶原纤维,有时胞核呈栅状排列。
(3)过度(混合)型:脑膜细胞与纤维细胞混合,排列成分叶状,中央为脑膜细胞,周围为纤维细胞,常形成旋涡状结构(图16-28),其中常包含有同心层状结构的砂粒体,乃变性肿瘤细胞及钙盐沉积。肿瘤质地硬,似砂砾状。
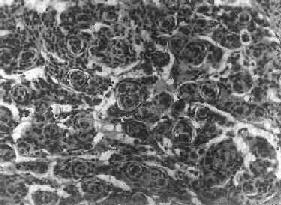
图16-28 脑膜瘤
瘤细胞为圆形或多角形,作旋涡状排列
(4)血管母细胞型:肿瘤细胞丰富,胞浆模糊,胞核椭圆,多排列在毛细血管旁,并有较多的网状纤维。
各型脑膜瘤中均可有不同程度的出血、钙化,有些并有黄色瘤细胞、软骨、骨、黑色素及粘液样变。
大多数脑膜瘤为良性,瘤细胞可引起邻近颅骨的骨质增生,或肿瘤浸润,但不引起广泛播散或转移,也不侵入邻近的神经组织。一般手术后复发率可达15%,其中血管母细胞型有复发和播散倾向,预后较差。少数脑膜瘤细胞间变明显,与梭形细胞肉瘤难以区分,可发生颅外转移,主要累及肺及淋巴结。
二、周围神经肿瘤
(一)神经鞘膜瘤
神经鞘膜肿瘤有神经鞘瘤(neurilemmoma)和神经纤维瘤(neurofibroma)两类。两者均为神经鞘细胞来源,但临床表现和组织形态有一定差别,故分别叙述。
1.神经鞘瘤神经鞘瘤可发生在周围神经、颅神经或交感神经。颅神经鞘瘤占颅内肿瘤5%~10%,发生于听神经,所以又称听神经瘤(acoustic neurinoma)因位于小脑脑桥角,故又称小脑脑桥角瘤(cerebello-pontine angle tumor),其次见于三叉神经。其他颅神经则极少受累,患者多为中、老年,女多于男(2:1)。
网眼观,神经鞘瘤有完整的包膜,大小不一,质实,呈圆形或结节状,常压迫邻近组织,但不发生浸润,与其所发生的神经粘连在一起。切面为灰白或灰黄色略透明,切面可见旋涡状结构,有时还有出血和囊性变。
镜下,肿瘤有二种组织形态。一型为束状型(Antoni A型),细胞细长,梭形,境界不清,核长椭圆形,互相紧密平行排列呈栅状或不完全的旋涡状,称Verocay小体(图16-29)。另一型为网状型(Antoni B型),细胞稀少,排列成稀疏的网状结构,细胞间有较多的液体,常有小囊腔形成。以上两型结构往往同时存在于同一肿瘤中,其间有过渡形式,但多数以其中一型为主。约10%病程较长的肿瘤,表现为细胞少,胶原纤维多,形成纤维瘢痕并发生玻璃样变,只在部分区域可见少量典型的神经鞘瘤的结构。
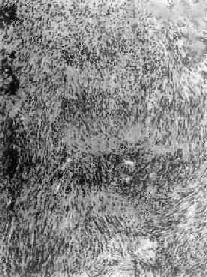
图16-29 神经鞘瘤
瘤细胞为长梭形,呈栅状排列
临床表现随其大小与部位而异,小肿瘤可无症状,较大者因受累神经受压而引起麻痹或疼痛,并沿神经放射。颅内听神经瘤可引起听觉障碍或耳鸣等症状。大多数肿瘤能手术根治,极少数与脑干或脊髓等紧密粘连未能完全切除者可复发,复发肿瘤仍属良性。
2.神经纤维瘤神经纤维瘤多发生在皮下,可单发也可多发性,多发性神经纤维瘤又称神经纤维瘤病(neurofibromatosis,von Recklinghausen’disease)。
肉眼观,皮肤及皮下单发性神经纤维瘤境界明显,无包膜,质实,切面灰白略透明,常不能找到其发源的神经。如发生肿瘤的神经粗大,则可见神经纤维消失于肿瘤中。肿瘤质实,切面可见旋涡状纤维,极少发生变性、囊腔形成或出血。
镜下,肿瘤由增生的神经鞘膜细胞和纤维母细胞构成,排列紧密,成小束并分散在神经纤维之间,伴多量网状纤维和胶原纤维及疏松的粘液样基质。
以上两型肿瘤均可发生恶变,但较多见于神经纤维瘤,尤其是神经纤维瘤病有较高的恶变倾向。表现为瘤细胞数目增多,出现多形性及核分裂像并伴有血管增生,肿瘤的形态颇似纤维肉瘤,称为神经纤维肉瘤。患者男多于女(4:1),从幼儿至老年均可发生,病程长,一般在5年以上。
(二)交感神经肿瘤
神经元性肿瘤中最原始而低分化的恶性肿瘤为神经母细胞瘤,高分化的良性肿瘤为节细胞神经瘤。这些肿瘤在中枢神经极为罕见,而较多发生在交感神经节与肾上腺髓质,详见肾上腺肿瘤。
三、转移性肿瘤
中枢神经系统的转移性肿瘤可累及脑和脊髓,其中以脑的转移性肿瘤较多见。在临床脑肿瘤中估计约有20%为转移性,而在恶性肿瘤死亡的病例中约有10%~15%有脑转移。
1.脑转移性肿瘤的来源 以支气管肺癌最常见,其中约40%发生脑转移,其次为乳腺癌约25%。其他肿瘤如胃癌、结肠癌、肾癌和黑色素瘤等均可发生脑转移,特别是黑色素瘤脑转移可达15%。
2.脑转移瘤的形成
(1)脑实质转移结节:转移性肿瘤结节界限清楚单个或多个,呈实性或部分囊性,内含粘液性物质、血性液体或坏死碎屑。
脑的转移性肿瘤多数位于皮质白质交界处,少数可位于脑的深部,各区域的转移率可能与其容积成正比。在白质中的转移肿瘤,周围水肿明显,有的甚为广泛,其程度与瘤结节大小不成比例。肿瘤的广泛出血常见于黑色素瘤、绒毛膜上皮癌和肺癌。
(2)软脑膜癌病(leptomeningeal carcinomatosis):又称癌性脑膜炎(carcinomatous meningitis)。肿瘤细胞发生弥漫性蛛网膜下腔浸润,以脑底部或脊髓背侧腰、骶、马尾部最为多见,同时累及其中的颅神经或脊髓神经根。肉眼观,病变轻微者,脑膜近乎正常或略混浊;严重者脑膜灰白,有大小不等之结节或斑块。由于蛛网膜下腔阻塞,脑积水明显。瘤细胞可循脑膜播散而累及脑室壁,通过血管周围间隙进而侵入脑实质。单纯的软脑膜癌病颇少见,多由于脑实质癌结节累及软脑膜所致。
(3)实质脑炎型转移:弥漫型血管周围瘤细胞浸润可形成局限性瘤结节或广泛浸润,常伴发软脑膜癌病。
3.转移性癌的组织形态与原发癌相似,癌组织有不同程度的坏死、液化、囊性变或出血。癌结节周边部常可见癌细胞沿血管周围浸润,脑组织水肿明显,有时并有变性坏死和淋巴细胞、巨噬细胞浸润及泡沫细胞形成。
第六节 脱髓鞘疾病
原发性脱髓鞘(primary demyelination)是指髓鞘脱失但轴索相对完好而言,乃由于少突胶质细胞受损影响髓鞘形成,或由于免疫或毒性因素损害髓鞘所致。继发性脱髓鞘常继发于轴突变性(如Waller变性)。脱髓鞘疾病是中枢神经系统以广泛原发性脱髓鞘为主的一组疾病,见表16-2。感染、缺氧和代谢障碍,以及其他一些先天性代谢障碍(如白质营养不良)、病毒感染(如进行性多灶性白质脑病)及某些原因不明的少见疾病(如脑桥中央白质溶解)时亦可出现脱髓鞘病变。
表16-2 脱髓鞘疾病分类
| 急性播散性脑脊髓炎(感染后性,疫苗接种后性,特发性) |
| 急性坏死出血性白质脑炎 |
| 多发性硬化症 |
| 经典型(Charcot病) |
| 大脑下白质广泛硬化症(Schilder病) |
| 同心圆硬化型(Balo病) |
| 视神经脊髓炎(Devic病) |
| 脱髓鞘伴全身性疾病 |
| 脑桥中央白质溶解 |
| 原发性胼胝体坏死 |
| 进行性多灶性白质脑病 |
一、多发性硬化症
多发性硬化症(multiple sclerosis,MS)是常见的脱髓鞘疾病,患者以20~40岁女性为多。临床病程数年至十余年不等。以反复发作与缓解交替为其特点,缓解期长短不一。神经系统的症状因累及部位不同而颇为多样。
【病理变化】
经典型MS病变分布广泛,可累及大脑、脑干、脊髓、视神经等处,其中以白质,特别以脑室角或室旁白质的病变最突出,但灰质也可受累。病灶呈圆形或不整形,大小不一,直径从0.1cm到数厘米不等,数目多少不一,新鲜病灶呈浅红色或半透明状,陈旧病灶呈灰白色,质地较硬。
镜下,脱髓鞘是本病的主要变化,早期多从静脉周围开始(又名静脉周脱髓鞘)伴血管周围单核细胞或淋巴细胞浸润。进行性脱髓鞘病灶的边缘常有多量单核细胞浸润,病灶中髓鞘变性崩解成颗粒状,并被吞噬细胞吞噬,形成泡沫细胞(图16-30)。轴索大多保存,部分可因变性而发生肿胀、扭曲断裂,甚至消失。此外,少突胶质细胞明显减少,甚至脱失;星形胶质细胞反应性增生十分明显,有时可出现肥胖细胞。晚期病灶胶质化,成为硬化斑。

图16-30 血管周围脱髓鞘(固蓝染色)
如脱髓鞘区与有髓鞘区相互交替,形成同心圆样结构,则称为同心圆性硬化又名Balo病,在我国东北和西南地区有散发病例的报道。近年观察发现,同心圆硬化和一般的脱髓鞘病灶可出现于同一病例;因此Balo病可能仅是经典型MS的某一阶段的表现。Schilder病则表现为大脑皮质下白质广泛的融合性脱髓鞘病变。皮质下弓状纤维的髓鞘保存完好是其特征。
部分病例病变主要累及脊髓和视神经,引起视力损害和脊髓症状,此即视神经脊髓炎又名Devic病,此型在远东常见。我国有仅累及脊髓的Devic病报道。
【病因和发病机制】
病因不明,可能和下列因素有关:①遗传因素:在欧美白人患者中HLA-A3,-B7和-DW2抗原阳性者较多。②人文地理因素:本病在寒温带多见,热带则较少。欧洲人发病率高,而东方、非洲人患病率较低。③感染因素:曾怀疑麻疹病毒、疱疹病毒和HIV病毒与本病有关,但即使应用分子生物学方法检测病灶内及周围脑组织中的病毒基因组,亦未能得出明确的结论。
动物实验表明,注射脑组织成分或狂犬病疫苗均可引起脱髓鞘病变,提示本病可能为多种因素诱发的变态反应疾病。在脱髓鞘病灶内可检出CD4+T(辅助)和CD8+T(抑制)细胞,然而确切的发病机制仍不清楚。
【临床病理联系】
本病病变分布广泛且轻重不等,故临床表现多样,有大脑、脑干、小脑、脊髓和视神经损害等症状,如肢体无力、感觉异常、痉挛性瘫痪、共济失调、眼肌麻痹、膀胱功能障碍等,病情发作和缓解可交替进行多年。
二、急性播散性脑脊髓炎
急性播散性脑脊髓炎可见于病毒(如麻疹、风疹、水痘等)感染后或疫苗(如牛痘疫苗、狂犬病疫苗等)接种后,临床表现为发热、呕吐、嗜睡、昏迷。一般在病毒感染后2~4天或疫苗接种后10~13天发病。
病变的特点为静脉周围脱髓鞘伴炎性水肿和以淋巴细胞和巨噬细胞为主的炎性细胞浸润。本病的脱髓鞘进展迅速,轴突一般不受累,病变呈多发性,累及脑和脊髓各处,特别是白质深层和脑桥腹侧。软脑膜中可有少量淋巴细胞、巨噬细胞浸润。
本病并非直接由病毒所致,在患者的中枢神经组织中不能检出病毒,加之病变与实验性过敏性脑脊髓炎十分相似,故目前认为本病髓鞘的损伤与髓鞘碱性蛋白所致的自身免疫反应有关。
三、急性坏死出血性白质脑炎
本病是一种罕见的发展迅速而凶险的疾病,常是败血性休克、过敏反应(哮喘等)的一种严重并发症。可能是一种由于免疫复合物沉积和补体激活所致的超急型急性播散性脑脊髓炎。病变的特点为脑肿胀伴白质点状出血(图16-31),与脑脂肪栓塞颇相似。镜下变化特点为小血管(小动脉、小静脉)局灶性坏死伴周围球形出血;血管周围脱髓鞘伴中性粒细胞、淋巴细胞、巨噬细胞浸润;脑水肿和软脑膜炎。与急性播散性脑脊髓炎之区别在于本病的坏死较广泛,急性炎性细胞浸润以及血管周围出血较明显。病变在大脑半球和脑干较多见,呈灶性分布。
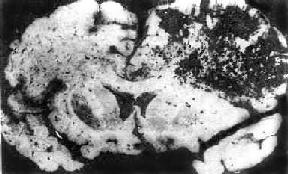
图16-31 急性坏死性出血性白质脑炎
大脑明显肿胀,白质中有许多点状出血
四、Guillian-Barre综合征
对称性多神经根炎(polyradiculoneuritis)或急性特发性多神经炎,是常见的脊神经根和周围神经的炎性脱髓鞘病变,不属原发性脱髓鞘疾病。临床上表现为进行性上升性麻痹、四肢软瘫、伴不同程度的感觉障碍。病变严重者,可引起致死性呼吸麻痹和两侧面瘫。脑脊液出现典型的蛋白细胞分离现象,即蛋白增加而细胞数正常。
本病可由多种病因引起,大多数病例在发病前先有诸如巨细胞病毒,EB病毒、支原体或HIV的感染,约20%病例的病因不明。本病的发生可能与免疫性损伤有关。以患者血清注射于动物的神经可产生静脉周围脱髓鞘病变。此外,患者的神经组织内有C3b免疫球蛋白(主要是IgG或IgM)存在。以上事实提示,本病的发生可能与体液免疫损伤有关。
病变可累及运动和感觉神经根、背根节及周围神经,主要表现为:①神经节和神经内膜水肿和灶性炎细胞浸润,②节段性脱髓鞘,崩解的髓鞘为巨噬细胞吞噬,③在严重病例,轴索可发生肿胀或断裂。轴索破坏严重时,相关的肌群可发生去神经性萎缩。在反复发作的慢性病例中,节段性脱髓鞘和受累神经纤维的修复过程反复进行,病变处神经鞘膜细胞突起与胶质纤维作同心圆状层层包绕,称为洋葱球形成。
第七节 变性疾病
变性疾病是一组原因不明的中枢神经系统疾病,病变特点在于选择性地累及某1~2个功能系统的神经细胞而引起受累部位特定的临床表现,如累及大脑皮层神经细胞的病变,主要表现为痴呆;累及基底核椎体外运动神经系统则引起运动障碍,累及小脑可导致共济失调等。本组疾病的共同病理特点为受累部位神经元的萎缩、死亡和星形胶质细胞增生,此外不同的疾病还可有各自特殊的病变,如在细胞内形成包含体或发生神经原纤维缠结等病变。几种主要的变性疾病见表16-3。
表16-3 几种主要的变性疾病
| 病变主要累及部位 | 疾 病 |
| 大脑皮质 | Alzheimer病 |
| Pick病 | |
| 基底核及脑干 | Huntington病 |
| 震颤性麻痹 | |
| 纹状体黑质变性 | |
| 进行性核上麻痹 | |
| Shy-Drager综合征 | |
| 脊髓与小脑 | 橄榄桥脑小脑萎缩 |
| Friedreich共济失调 | |
| 共济失调性毛细血管扩张症 | |
| 运动神经元 | 肌萎缩性侧索硬化 |
| 脊髓性肌萎缩 |
一、Alzheimer病
Alzheimer病又称初老期痴呆,是以进行性痴呆为主要临床表现的大脑变性性疾病,起病多在50岁以后。随着人类寿命的延长,本病的发病率呈增高趋势。按照美国的诊断标准,上海60岁以上人群发病率为3.46%。65岁以上人群为4.61%。临床表现为进行性精神状态衰变,包括记忆、智力、定向、判断能力、情感障碍和行为失常甚至发生意识模糊等。患者通常在发病后5~6年内死于继发感染和全身衰竭。
【病理变化】
肉眼观,脑萎缩明显,脑回窄、脑沟宽,病变以额叶、顶叶及颞叶最显着(图16-32),脑切面可见代偿性脑室扩张。
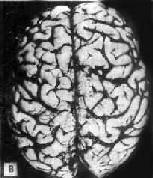
图16-32 初老期痴呆的脑
示脑明显萎缩(A),与正常脑(B)的对比
镜下,本病最主要的组织病变有:老年斑,神经原纤维缠结,颗粒空泡变性,Hirano小体等。
(1)老年斑:为细胞外结构,直径为20~150μm,最多见于内嗅区皮质、海马CA-1区,其次为额叶和顶叶皮质。银染色显示,斑块中心为一均匀的嗜银团,刚果红染色呈阳性反应,提示其中含淀粉样蛋白,其中含该蛋白的前体β/A-4蛋白及免疫球蛋白成分。中心周围有空晕环绕,外围有不规则嗜银颗粒或丝状物质。电镜下可见该斑块主要由多个异常扩张变性之轴索突触终末构成(图16-33)。
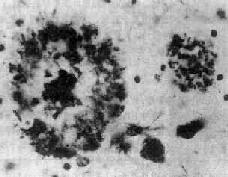
图16-33 老年斑
左侧为典型的老年斑。中心为嗜银团,围以空晕,外围为嗜银性细颗粒及细丝,周围见星形胶质细胞。右侧见一正在形成的老年斑
(2)神经原纤维缠结:神经原纤维增粗扭曲形成缠结,在HE染色中往往较模糊,呈淡蓝色,而银染色最为清楚。电镜下证实其为双螺旋缠绕的细丝构成,多见于较大的神经元,尤以海马、杏仁核、颞叶内侧,额叶皮质的锥体细胞最为多见。此外,前脑底Meynert基底核及蓝斑中也可见到。这一变化是神经元趋向死亡的标志(图16-34)。
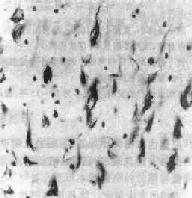
图16-34 神经原纤维缠结
脑皮质锥体细胞神经原纤维缠结增粗呈焰状(Bielschowsky银染色)
(3)颗粒空泡变性:表现为神经细胞胞浆中出现小空泡,内含嗜银颗粒,多见于海马Sommer区的锥体细胞。
(4)Hirano小体:为神经细胞树突近端棒形嗜酸性包含体,生化分析证实大多为肌动蛋白,多见于海马锥体细胞。
上述变化均非特异性,可见于无特殊病变之老龄脑,仅当其数目多并具特定的分布部位时才能作为Alzheimer病的诊断依据。
【病因和发病机制】
病因和发病机制不明。对于本病究竟是一独立的疾病,还是一种加速的老化尚有不少争议。本病的发病可能与下列因素有关:①受教育程度:上海的人群调查资料以及随后世界大多数地区的调查资料证实,本病的发病率与受教育程度有关。文盲及初小文化人群中发病率最高,受到高中以上教育人群中发病率较低。病理研究表明,大脑皮质突触的丧失先于神经元的丧失,突触丧失的程度和痴呆的相关性较老年斑、神经原纤维缠结与痴呆的相关性更加明显。人的不断学习可促进突触的改建,防止突触的丢失。②遗传因素:对初老期痴呆病中具有家属遗传史的病人(遗传性Alzheimer病仅为本病一个少见类型)的研究表明,其控制基因在第21对染色体上,具有多个位点,某些位点与先天愚型(Down症)位点甚接近,因此后者的Alzheimer病的发病率较高。③神经细胞的代谢改变:老年斑中的淀粉样蛋白的前体β/A-4蛋白是正常神经元膜上的一个跨膜蛋白,何以在本病中会发生不溶性沉淀的原因尚待探讨。缠结的神经原纤维中神经微丝,τ蛋白等细胞骨架蛋白呈现过度的磷酸化。某些患者病脑中铝的含量可高于正常。④继发性的递质改变:其中最主要的改变是乙酰胆碱的减少。由于Meynert基底核神经元的大量缺失致其投射到新皮质、海马、杏仁核等区域的乙酰胆碱能纤维减少。综上所述,目前已发现了本病的形态、生化、遗传等各方面的异常改变,但病因和发病机制尚有待阐明。
病理诊断
由于Alzheimer病的病理变化均为非特异性,故必须根据老年斑和神经原纤维缠结的数目及部位,结合患者的年龄和临床表现才能作出判断。参考Khachaturian执笔的美国约定诊断标准(表16-4),并除外其它引起痴呆的原因,如血管源性痴呆以及其它变性疾病等,方可作出诊断。
表16-4 美国诊断Alzheimer病的标准
| 患者年龄 | 老年斑数(×200,/mm2) | 神经原纤维缠结(×200,/mm2) |
| <50岁 | >2~5 | >2~5 |
| ≤60 | >8 | >2~5 |
| ≤75 | >10 | >2~5 |
| >75 | >15 | 有或无 |
二、慢性进行性舞蹈病
慢性进行性舞蹈病又称Huntington舞蹈病,是一种常染色体显性遗传病,突变基因位于第4对染色体。患者的子女中半数可得病,男女患病机会均等,多在20~50岁开始发病。临床表现为舞蹈样动作及进行性痴呆。
脑明显缩小,重量小于1000g,最突出的是两侧尾状核和壳核的萎缩,以致侧脑室明显扩张。大脑皮质特别是额、顶叶萎缩显着,白质也减少。
镜下可见尾状核和壳核中选择性小神经细胞丢失,伴星形胶质细胞增生和胶质纤维化,类似的病变可见于丘脑腹侧核和黑质。
本病呈进行性发展,病程多为10~15年,最后死于并发症。
三、震颤性麻痹
震颤性麻痹(paralysis agitans)又称Parkinson病,是一种缓慢进行性疾病,多发生在50~80岁。临床表现为震颤、肌强直、运动减少、姿势及步态不稳、起步及止步困难、假面具样面容等。
本病的发生与纹状体黑质多巴胺系统损害有关,最主要的是原因不明性(特发性)Parkinson病,其他如甲型脑炎后,动脉硬化,及一氧化碳、锰、汞中毒等,均可产生类似震颤性麻痹症状或病理改变。这些情况统称为Parkinson综合征。
【病理变化】
黑质和蓝斑脱色是本病肉眼变化的特点(图16-35)。镜下可见该处的黑色素细胞丧失,残留的神经细胞中有Lewy小体形成,该小体位于胞浆内,呈圆形,中心嗜酸性着色,折光性强,边缘着色浅。电镜下,该小体由细丝构成,中心细丝包捆致密,周围则较松散。
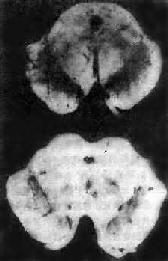
图16-35 Parkinson病
中脑黑质脱色(下),正常中脑黑质完好(上)
由于黑质细胞的变性和脱失,多巴胺合成减少,以致多巴胺(抑制性)与乙酰胆碱(兴奋性)的平衡失调而致病。近年来用左旋多巴(多巴胺的前体)来补充脑组织中多巴胺不足或用抗胆碱能药物以抑制乙酰胆碱的作用,对本病有一定的疗效。
四、肌萎缩性侧索硬化症
肌萎缩性侧索硬化症(amyotrophic lateral sclerosis,ALS)的病变累及锥体束上、下神经元,上神经元在大脑皮质,其轴索经过内囊、脑干及皮质脊髓束与脑运动神经核或脊髓前角下运动神经元相联系。本病发病年龄在40~50岁,男多于女,临床起病缓慢,表现为进行性上、下肢肌萎缩,无力,锥体束损害及脑干运动神经核受损,一般无感觉障碍,病程一般2~6年。
本病的病变多自脊髓向上发展,波及脑干乃至大脑的运动区。下运动神经元变性消失,以颈、腰膨大区最为显着,脊髓前根及其支配的肌肉萎缩。上运动神经元大量变性消失伴星形胶质细胞增生。肉眼观,中央前回萎缩明显。在延髓麻痹型病例中,脑运动神经核特别是第Ⅴ、Ⅸ、Ⅹ、Ⅺ及Ⅻ神经核的神经细胞变性脱失,偶见神经细胞被吞噬,提示持续性神经细胞丢失。残留的神经元皱缩成鬼影状。
运动神经元病的病因和发病机制尚不清楚。野生小鼠自发性运动神经元病系由C型RNA病毒引起,但与人类疾病有无关系尚不清楚。值得注意的是关岛Chamorro族和日本纪伊半岛中本病的发病率高,该两地区土壤中含锰甚高,饮水中钙的含量也高,尸检病例脊髓中含锰量也甚高,因而锰、钙在发病中的作用已受到注意。
第八节 代谢性疾病
代谢性疾病累及神经系统者有两类。
一、代谢性脑病
本病是系统性疾病在脑的表现,由于血脑屏障发生障碍,脑组织受生化内环境的影响,发生代谢变化,导致脑功能障碍。常见的病因有:糖尿病、尿毒症、高血钙症及肝功能衰竭等。往往脑功能障碍显着,但病理形态变化不明显。提示本型脑病的性质主要是生化性障碍。例如:
肝性脑病(hepatic encephalopathy)本病是严重肝病引起的中枢神经系统的综合征。临床上以扑击样震颤、精神和行为改变、意识障碍终至昏迷为主要表现。由于本病晚期常出现昏迷,又称为肝昏迷(hepatic coma)。
本病的发病机制甚为复杂,表现为下列诸方面的代谢紊乱:①氨中毒与供能衰竭。肝硬变时由于门腔静脉的侧枝循环分流,肠道内的氨(胺)类物质未经肝解毒而直接进入中枢神经系统。在星形胶质细胞和神经元中氨与α-酮戊二酸生成谷氨酸,后者与氨又生成谷氨酰胺。在此过程中消耗了大量的ATP和α-酮戊二酸,使细胞不能维持糖的正常有氧代谢和能量供应。神经细胞中的谷氨酸除可形成谷氨酰胺外还可在谷氨酸脱羧酶的作用下形成抑制性递质γ-氨基丁酸(GABA)。此外过多的氨还可抑制丙酮酸脱氢酶的活力、抑制Na-K-ATP酶活力、改变Na+、K+在神经细胞膜上的正常分布,从而干扰神经传导活动。②递质紊乱。包括原有递质的失衡和伪递质的产生。食物中的芳香氨基酸经肠道细菌脱羧后形成酪胺和苯乙胺。它们进入神经系统后,在脑内经β-羟化酶作用,分别形成鱆胺(octopamine)和苯乙醇胺。两者的结构与去甲肾上腺素和多巴胺很相似,但传递神经冲动功能仅为原递质的十分之一。去甲肾上腺素和多巴胺为兴奋性递质,如兴奋冲动不能传递,则可出现意识障碍和昏迷。此外,多巴胺被替代后,锥体外系的乙酰胆碱递质占优势,乃出现扑击样震颤。如前所述由于解氨毒的结果抑制性递质GABA增多,与肝昏迷也有一定关系。③其它因素如短链脂肪酸增多,胰岛素——血浆氨基酸失衡,低血糖等因素对肝昏迷发生也起了一定的作用。
肝性脑病患者约有半数可出现不同程度的脑水肿。慢性肝性脑病患者的主要病变见于大脑和小脑的灰质、基底核、丘脑、红核和黑质。这些部位的星形胶质细胞增生,神经细胞无明显形态改变。增生的星形胶质细胞胞浆不明显,核大扭曲,核膜增厚,核浆内含有糖原(PAS染色呈阳性反应),HE染色显得核浆苍白,并有1~2个明显核仁,此种细胞称为AlzheimerⅡ型细胞(图16-36),其GFAP染色则从阳性转为阴性,反映了此类细胞在解氨毒过程中其自身代谢发生了紊乱。这种在自身代谢紊乱基础上发生的细胞增生称为营养不良性星形胶质细胞增生(dystrophic astrogliosis)。长期严重的肝性脑病还可导致神经元和神经纤维丧失,大脑皮质变薄,皮质深部层状坏死,可累及大脑、小脑和基底核。
图16-36 AlzheimerⅡ型细胞
该细胞核大,核浆苍白,核仁明显,核膜增厚扭曲,呈典型“空泡状”核
二、先天性代谢障碍
先天性代谢障碍常导致多系统受累。临床上有些以神经系统表现为主,有些则以神经系统外的表现为突出:如肝豆状核变性(Wilson病)患者中,有的主要表现为肝功能障碍,神经系统症状轻微;而另一些患者则有突出的神经系统症状,表现为舞蹈征、手足徐动或痴呆。又如苯丙酮尿症(phenylketonuria)患者,代谢障碍遍及全身各组织,但临床上神经系统的症状特别显着。值得注意的是同一种代谢障碍可产生不同的临床表现,而不同的代谢障碍也可产生相同的临床现象,故本组疾病至今尚无明确的分类。属于先天性代谢障碍病者尚有系统性贮积病(如鞘氨醇脂质沉积症、Gaucher病),神经元贮积病(如Tay-sack病,Niemann-Pick病),白质营养不良等。其他累及神经系统者尚有粘多糖沉积症,糖原沉积症等。现以白质营养不良为例,简述如下:
白质营养不良症(leukodystrophy)是一种遗传性神经鞘磷脂代谢障碍而影响髓鞘形成的疾病。包括异染色性白质营养不良、球样细胞白质营养不良、肾上腺白质营养不良、Alexander病、海绵状脑白质营养不良和Pelizaeus-Merzbacher病等。本组疾病多见于婴儿及儿童,预后不良。其中以异染性白质营养不良和球样细胞白质营养不良较为常见。
(一)异染性白质营养不良
异染性白质营养不良(metachromatic leukodystrophy)本病为常染色体隐性遗传病,由于硫酸脂酶缺乏使硫酸脑苷酯沉积于中枢神经系统的白质和周围神经中。以甲苯胺蓝或结晶紫染色,不呈紫色而呈黄褐色,具有异染性,故而得名。脑外形与重量大致正常,切面见白质灰白色,质硬,皮质下弓状纤维不被累及。镜下见白质中髓鞘广泛形成不良,轴突变性,星形胶质细胞增生,少突胶质细胞减少,可见异染性脂质沉积于神经元和巨噬细胞胞浆中。视网膜、视神经和周围神经常被严重累及,Schwann细胞、肝枯否细胞和肾小管上皮细胞也可有异染性脂滴存在。
(二)球样细胞白质营养不良
球样细胞白质营养不良(globoid leukodystrophy) 又称Krabbe病,为常染色体隐性遗传病,由于β-半乳糖脑苷酶缺乏致使β-半乳糖脑苷沉积。病变表现为大脑和脊髓白质广泛髓鞘形成不良,但皮质下弓状纤维不被累及。在白质中出现特征性的上皮样及球样细胞,后者直径20~40μm,胞浆中的半乳糖脑苷呈PAS染色阳性,苏丹黑染色弱阳性。电镜观察可见该细胞含有空管状包含体,其中有不规则结晶。此外可见局灶性神经元丢失,轴索消失和反应性星形胶质细胞增生。周围神经轻度灶性髓鞘形成不良,Schwann细胞和巨噬细胞含有PAS阳性物质,但很少形成典型球样细胞。
第十七章 骨关节疾病
骨关节疾病种类很多,包括发育障碍、损伤,炎症、代谢和内分泌紊乱以及肿瘤和瘤样病变等。本章主要介绍骨折愈合、常见骨肿瘤、佝偻病和骨软化症、类风湿关节炎和大骨节病。为了便于理解本章的内容,兹复习骨和关节的基本结构,着重于与病变有关的内容。
骨是一种特殊的结缔组织,由多种细胞和细胞间的骨基质组成。
1.骨基质(bone matrix)由有机物质和无机盐构成。包括:①胶原纤维,约占90%,主要是Ⅰ型胶原;②蛋白多糖(proteoglycan),构成胶原纤维间的无定形物质;③骨盐(bony salt)主要是羟磷灰石。
2.骨的细胞成分 包括:①骨母细胞(osteoblast):胞浆内有丰富的粗面内质网和核蛋白体,合成前胶原蛋白,经高尔基器加工后,释出细胞外,进一步形成胶原纤维。另外,还合成和分泌蛋白多糖。一旦有机骨基质形成,钙化亦即开始。骨母细胞中有大量线粒体,其基质中含有丰富的钙磷颗粒(焦磷酸锑钙)。骨母细胞富于碱性磷酸酶,具有焦磷酸酶的活性,可以水解焦磷酸盐,还可水解磷酸酯,提高局部钙和磷酸盐的浓度,形成过饱和状态,而促进钙化过程。电镜下,骨母细胞以分泌基质小泡的方式,释放其内容物,使有机骨基质钙化。近年发现骨母细胞合成骨钙蛋白(osteocalcin)和骨联蛋白(osteonectin),它们是维生素K依赖性钙结合蛋白,能促使矿物盐与胶原纤维的结合。骨母细胞有维生素D受体,还有甲状旁腺激素受体以及胶原酶和激活胶原酶等的血浆素原活素(plasminogen activator),表明它也可能参与骨质吸收。骨母细胞的大小、形态与其功能有关,在骨形成活跃时骨母细胞肥硕、呈立方或柱状;当骨形成静止时则变为扁平或梭形。②骨细胞(osteocyte):骨母细胞分泌骨基质的过程中自身被包埋在其陷窝之中而成为骨细胞。骨细胞除参与骨的合成外,也可能参与骨的吸收,往往可见陷窝周围有骨质溶解。③破骨细胞(osteoclast):来自单核巨噬细胞,为一种多核巨细胞,含有丰富的酸性磷酸酶、胶原酶和蛋白多糖酶等,具有吸收内和钙化软骨的功能。电镜下,破骨细胞的细胞膜在贴近被吸收骨的一侧形成密集的皱褶,称为皱褶缘(ruffled border),以增加骨质吸收的表面积。破骨细胞的细胞膜上有甲状旁腺激素和降钙素的受体,用以调节其功能。人的淋巴细胞可以产生破骨细胞激活因子(OAF),可诱导破骨细胞的形成和吸收骨质,前列腺素也可引起骨质吸收。
【骨的形成和吸收】
1.骨的形成通过膜内化骨和软骨内化骨两种方式完成,两者均需经过两个阶段,即先形成有机骨质,称为类骨(osteoid),继而钙化为骨质。现以长骨为例,说明上述二种骨形成方式。
长骨由骨干和两端的骨骺端组成,在骨干的近骨骺处称为干骺端。在青春期末前,骨骺和骨干之间仍保留一层软骨板,称为骺板。在骺板软骨与干骺端之间有软骨化骨的过程,这是长骨增长的基础。软骨化骨是一个连续的过程,形态上分为四个区,即静止软骨区、增生软骨区,软骨基质钙化区和骨化区。在骺板近骨骺处软骨细胞依骨的长轴排列呈柱状,并不断增生和分泌软骨基质,而在近干骺端处软骨基质不断钙化,软骨细胞不断退化、死亡,并由破骨细胞吸收清除。同时血管及骨母细胞侵入,在正在清除的软骨基质表面有整齐排列成行的新生骨母细胞,不断形成类骨质,并进一步钙化形成骨质。青春期后(约17~22岁)骺板完全骨化,长骨停止生长,该处留有一条线形痕迹,称为骺线。骨内、外表面均被覆一层骨膜。骨外膜由致密的结缔组织构成,其深层的间叶细胞可分化为骨母细胞形成类骨和骨质,这种骨形成过程称膜内化骨。骨内膜被覆骨髓腔面和骨小梁表面,为薄层的结缔组织,间叶细胞排列呈单层如扁平上皮,亦能分化为骨母细胞,骨内膜还有破骨细胞。
2.骨的吸收矿物质和有机骨基质成分是同时被吸收的,主要由破骨细胞完成。被吸收的骨质表面形成凹陷,也称为Howship陷窝。类骨组织具有较强的抵抗力,不被破骨细胞吸收。
在整个生命过程中,骨组织在不断更新,骨的形成和吸收不断进行。幼年时新骨形成大于吸收,成人两者趋于平衡,老年人则骨质吸收明显大于形成,骨质日益疏松。
【骨的类型】
由于骨基质的胶原纤维和骨细胞排列不同,人类骨质在镜下可分为两种类型。
1.编织骨(woven bone)骨基质的胶原纤维粗短,呈不规则或无定向排列,骨陷窝及骨细胞均较大且排列紧密。编织骨属于非永久性骨,见于生长旺盛的骨质如胚胎或4岁以下的幼儿时期,经过一段时期后转变为板层骨。在病理情况下见于骨折后形成的骨痂及炎症病变部位之新生骨,也可见于骨肿瘤及瘤样病变如骨肉瘤、纤维结构不良等。
2.板层骨(lamellar bone)骨基质的胶原纤维较纤细,排列规则,有一定方向,如在松质骨其排列与骨小梁的长轴平行,而在致密骨或皮质骨则围绕血管呈同心圆排列(Havers系统)。用嗜银染色或偏光显微镜观察则十分明显。
关节可分为不动关节(synarthroses)和动关节(diarthroses)。动关节又称滑膜性关节,由关节软骨、滑膜以及关节囊和周围的韧带、肌肉组成。动关节容易发生病变:
1.滑膜(synovial membrane) 衬于关节囊内面并附于关节软骨周缘,有内、外两层,内层为被覆于关节腔的滑膜细胞(synoviocyte),形成不连续的单层扁平细胞或一到多层梭形、椭圆形或立方形细胞层。滑膜细胞直接与外层相连,外层为富于血管和淋巴管的疏松结缔组织。与浆膜腔不同,两者之间无基膜,有利于滑液和血管、淋巴管之间的物质渗透和交换。炎症时也容易造成病变扩散蔓延。正常的滑膜平滑,可形成小绒毛状突起或含脂肪组织的皱襞,充填关节内的空隙。
电镜下,滑膜细胞分两种:一种为A型或巨噬细胞样细胞(M)型,胞浆内有大量溶酶体和吞噬空泡,多丝状伪足。主要吞噬进入关节的内源性或外来异物,还合成降解酶,主要是胶原酶;另一种为B型或纤维母细胞样细胞(F)型,胞浆内有丰富的粗面内质网和核蛋白体,主要合成滑液内透明质酸盐-蛋白质。滑膜细胞再生能力强,滑膜血管外膜细胞可化生为滑膜细胞。滑膜受损后易于修复,且可过度增生,形成各种形状的绒毛甚至结节(如类风湿关节炎、绒毛结节性滑膜炎)。偶尔可化生形成软骨或骨化小灶(如滑膜软骨瘤病)。
关节腔内含有滑液,滑液由血浆透析而来,再加以滑膜细胞合成的透明质酸盐-蛋白质,而增加了粘稠度。滑液的作用是润滑关节面及营养关节软骨。
2.关节软骨为透明软骨,表面光滑,柔软而富有弹性。由软骨细胞及软骨基质组成。软骨细胞分三层:表层为梭形,其细胞长轴与表面平行;中层呈小柱状排列,细胞可分裂增生;深层软骨细胞肥大,排列紧密,与其下的骨髓以狭窄的钙化软骨基质相分隔。软骨基质由软骨细胞产生和分泌,主要由Ⅱ型胶原纤维和蛋白多糖组成。Ⅱ型胶原纤维束粗大而致密交织成坚固的网架,保持软骨结构的完整性。蛋白多糖主要为硫酸软骨素A和C和硫酸角质素。它们是由一个核心蛋白连结许多氨基多糖侧链所组成,有像瓶刷状的空间分子结构,这种高度亲水性分子结构使关节软骨保持弹性,关节软骨本身无血管和淋巴管,其营养来自滑液,这种分子结构以及关节的活动可促进两者之间的物质交换。蛋白多糖又是多阴离子的大分子,可与带正电荷的胶原分子形成非可溶性复合物,亦可与各种染料结合。HE染色呈淡蓝色,爱先蓝染色阳性呈蓝色,甲苯胺蓝呈异染反应(紫红色)。病变时由于基质蛋白多糖降解,软骨染色反应可发生改变。HE呈红色,异染反应也消失。软骨细胞可合成胶原酶等蛋白溶解酶,与病变时软骨的降解有关。
第一节 骨折愈合
骨折(fracture of bone)通常可分为外伤性骨折和病理性骨折两大类。骨的再生能力很强,经过良好复位后的外伤性骨折,一般在3~4个月或更长一些时间内,可完全愈合。骨外、内膜中骨母细胞的增生和产生新生骨质是骨折愈合的基础。骨折后经血肿形成、纤维性和骨性骨痂形成以及骨痂改建的过程而完全愈合,使骨在结构和功能上恢复正常。
一、骨折愈合过程
骨折愈合(fracture healing)过程可分为以下几个阶段:
(一)血肿形成
骨折时除骨组织被破坏外,也一定伴有附近软组织的损伤或撕裂。骨组织和骨髓都富含血管,骨折后常伴有大量出血,填充在骨折的两断端及其周围组织间,形成血肿。一般在数小时内血肿发生血液凝固。和其他组织的创伤一样,此时在骨折局部还可见轻度中性粒细胞浸润。
骨折时由于骨折处营养骨髓、骨皮质及骨膜的血管随之发生断裂,因此在骨折发生的1~2天内,可见到骨髓造血细胞的坏死,骨髓内脂肪的析出,以后被异物巨细胞包绕形成脂肪“囊”(fat“cyst”)。骨皮质亦可发生广泛性缺血性坏死,骨坏死在镜下表现为骨陷窝内的骨细胞消失而变为空穴。如果骨坏死范围不大,可被破骨细胞吸收,有时死骨可脱落、游离而形成死骨片。
(二)纤维性骨痂形成
大约在骨折后的2~3天,从骨内膜及骨外膜增生的纤维母细胞及新生毛细血管侵入血肿,血肿开始机化。这些纤维母细胞实质上多数是软骨母细胞及骨母细胞的前身。上述增生的组织逐渐弥合,填充并桥接了骨折的断端,继而发生纤维化形成纤维性骨痂,或称暂时性骨痂(provisional callus)肉眼上骨折局部呈梭形肿胀。约经1周左右,上述增生的肉芽组织及纤维组织部分可进一步分化,形成透明软骨。透明软骨的形成一般多见于骨外膜的骨痂区,而少见于骨髓内骨痂区,可能与前者血液供应较缺乏有关。此外,也与骨折断端的活动度及承受应力过大有关。但当骨痂内有过多的软骨形成时会延缓骨折的愈合时间。
(三)骨性骨痂形成
骨折愈合过程的进一步发展,是骨母细胞产生新生骨质逐渐取代上述纤维性骨痂。开始形成的骨质为类骨组织(图17-1),以后发生钙盐沉着,形成编织骨(woven bone),即骨性骨痂(图17-2)。纤维性骨痂内的软骨组织,和骨发育时的软骨化骨一样,发生钙盐沉着而演变为骨组织,参与骨性骨痂的形成。此时所形成的编织骨,由于其结构不够致密,骨小梁排列比较紊乱,故仍达不到正常功能需要。
图17-1 骨痂
左侧为软骨性骨痂,右侧为类骨组织
图17-2 骨痂
在幼稚编织骨骨小梁表面的骨母细胞排列成行
按照骨痂的细胞来源及骨痂的部位不同,可将骨痂分为外骨痂和内骨痂。
1.外骨痂(external callus)或骨外膜骨痂(periosteal callus),是由骨外膜的内层即成骨层细胞增生,形成梭形套状,包绕骨折断端。如上所述,以后这些细胞主要分化为骨母细胞形成骨性骨痂,但也可分化为软骨母细胞,形成软骨性骨痂。在长骨骨折时以外骨痂形成为主。
2.内骨痂(internal callus)由骨内膜细胞及骨髓未分化间叶细胞演变成为骨母细胞,形成编织骨。内骨痂内也可有软骨形成,但数量比外骨痂为少。
(四)骨痂改建或再塑
上述骨痂建成后,骨折的断端仅被幼稚的、排列不规则的编织骨连接起来。为了符合人体生理要求而具有更牢固的结构和功能,编织骨进一步改建成为成熟的板层骨,皮质骨和髓腔的正常关系也重新恢复。改建是在破骨细胞的骨质吸收及骨母细胞新骨质形成的协调作用下进行的,即骨折骨所承受应力最大部位有更多的新骨形成而机械性功能不术需要的骨质则被吸收,这样就使骨折处上下两断端按原来的关系再连接起来,髓腔也再通。
在一般情况下,经过上述步骤,骨折部恢复到与原来骨组织一样的结构,达到完全愈合。
二、影响骨折愈合的因素
1.全身性因素①年龄:儿童骨组织再生能力强,故骨折愈合快;老年人骨再生能力较弱,故骨折愈合时间也较长。②营养:严重蛋白质缺乏和维生素C缺乏可影响骨基质的胶原合成;维生素D缺乏可影响骨痂钙化,妨碍骨折愈合。
2.局部因素①局部血液供应:如果骨折部血液供应好则骨折愈合快,如肱骨的外科颈(上端)骨折;反之,局部血液供应差者,骨折愈合慢,如股骨颈骨折。骨折类型也和血液供应有关:如螺旋形或斜形骨折,由于骨折部分与周围组织接触面大,因而有较大的毛细血管分布区域供应血液,愈合较横形骨折快。②骨折断端的状态:骨折断端对位不好或断端之间有软组织嵌塞等都会使愈合延缓甚至不能接合。此外,如果骨组织损伤过重(如粉碎性骨折),尤其骨膜破坏过多时,则骨的再生也较困难。骨折局部如出血过多,血肿巨大,不但影响断面的接触,且血肿机化时间的延长也影响骨折愈合。③骨折断端的固定:断端活动不仅可引起出血及软组织损伤,而且常常只形成纤维性骨痂而难有新骨形成。为了促进骨折愈合,良好的复位及固定是必要的。但长期固定可引起骨及肌肉的废用性萎缩,也会影响骨折愈合。④感染:开放性骨折(即骨折处皮肤及软组织均断裂,骨折处暴露)时常合并化脓性感染,延缓骨折愈合。
骨折愈合障碍者,有时新骨形成过多,形成赘生骨痂,愈合后有明显的骨变形,影响功能的恢复。有时纤维性骨痂不能变成骨性骨痂并出现裂隙,骨折两断端仍能活动,形成假关节,甚至在断端有新生软骨被覆,形成新关节。
附 病理性骨折
病理性骨折(pathological fracture)是指已有病变的骨,在通常不足以引起骨折的外力作用下发生的骨折,或没有任何外力而发生的自发性骨折。
常见的病理性骨折的原因:
1.骨的原发性或转移性肿瘤是病理性骨折最常见的原因,特别是溶骨性的原发或转移性骨肿瘤。原发性骨肿瘤如多发性骨髓瘤、骨巨细胞瘤及溶骨性成骨肉瘤等;属于转移性骨肿瘤的如转移性肾癌、乳腺癌、肺癌、甲状腺癌及神经母细胞瘤等。不少原发性和转移性骨肿瘤有时因病理性骨折后才被发现。
2.骨质疏松(osteoporosis)老年、各种营养不良和内分泌等因素可引起全身性骨质疏松,表现为骨皮质萎缩变薄,骨小梁变细、数量减少。主要影响脊椎骨、股骨颈、掌骨等。老年尤其是绝经后老年妇女胸、腰椎压缩性骨折,股骨颈、肱骨上端及桡骨下端骨折较为多见。肢体瘫痪、长期固定或久病卧床等可引起局部废用性骨质疏松而造成骨折。
3.内分泌紊乱由甲状旁腺腺瘤或增生引起的甲状旁腺功能亢进,可导致骨的脱钙及大量破骨细胞堆积,骨小梁为纤维组织所取代。此时虽有新骨形成,但只能形成纤细的编织骨或非钙化的类骨组织,而极易发生多发性病理性骨折。
4.骨的发育障碍有多种属于这类的先天性骨疾患可以引起病理性骨折。例如先天性成骨不全(osteogenesis imperfecta),为一种常染色体显性遗传性疾病,在胎儿或儿童时期发病,乃由于先天性间充质发育缺陷,不易分化为骨母细胞,同时骨母细胞合成骨基质中Ⅰ型胶原纤维障碍,因此长骨骨皮质很薄,骨细而脆,极易发生多发性病理性骨折,故又称为脆性骨综合征(brittle bone syndrome)。而骨折后新形成的骨痂为软骨性,或为纤维性,难以发生骨化。
病理性骨折时,骨的原有病变往往使骨折愈合迟缓,甚至几乎没有修复反应。也常使骨原有病变的组织学图像发生改变或复杂化。
第二节 骨肿瘤
骨肿瘤有原发性及继发性两大类。从发病率来看,后者比前者多见。恶性原发性骨肿瘤约占人体全部恶性肿瘤的1%,虽较为少见,但多发生于青壮年,在大范围手术治疗(如截肢、关节解脱术)后,不仅致残,而且仍有较高的死亡率,因此早期诊断及早期治疗十分重要。
一、原发性骨肿瘤的分类
原发性骨肿瘤通常以病理形态为基础,主要是根据肿瘤细胞的形态及其所产生的基质,再结合临床及X线改变,进行分类。根据肿瘤的分化程度和生物学特性的不同,又可将每类骨肿瘤区分为良性和恶性两大类(表17-1)。骨的瘤样病变本质上不是真性肿瘤,但因其在临床X线或病理形态上均与骨肿瘤相似,易与骨肿瘤混淆,故常在骨肿瘤中予以叙述。
表17-1 常见骨肿瘤分类
| 分 类 | 良 性 | 恶 性 |
| 成骨性肿瘤 | 骨瘤、骨样骨瘤、良性骨母细胞瘤 | 骨肉瘤、皮质旁骨肉瘤、恶性骨母细胞瘤 |
| 成软骨性肿瘤 | 骨软骨瘤(单发、多发)、软骨瘤(单发、多发)、良性软骨母细胞瘤、软骨粘液样纤维瘤 | 软骨肉瘤、恶性软骨母细胞瘤、间叶性软骨肉瘤、去分化软骨肉瘤 |
| 多核巨细胞 | 良性骨巨细胞瘤 | 恶性骨巨细胞瘤 |
| 骨髓源性肿瘤 | Ewing肉瘤、骨髓瘤、恶性淋巴瘤 | |
| 结缔组织性肿瘤 | 韧带样纤维瘤、非骨化性纤维瘤 | 纤维肉瘤 |
| 脉管组织性肿瘤 | 血管瘤、淋巴管瘤、血管球瘤 | 血管内皮瘤、血管外皮瘤 |
| 脂肪组织性肿瘤 | 脂肪瘤 | 脂肪肉瘤 |
| 神经组织性肿瘤 | 神经鞘瘤、神经纤维瘤 | 恶性神经鞘瘤 |
| 脊索源性肿瘤 | 脊索瘤 | |
| 瘤样病变 | 孤立性骨囊肿、动脉瘤性骨囊肿、骨的纤维结构不良、嗜酸性肉芽肿 |
二、常见的骨肿瘤
(一)骨软骨瘤
骨软骨瘤(osteochondroma)又名骨软骨外生性骨疣(osteocartilaginous exostosis),是指发生在骨表面,表面覆以软骨帽的疣状骨性隆起。骨软骨瘤是最常见的骨肿瘤,约占良性骨肿瘤的1/3。本瘤多见于11~20岁,约占40%。有单发性(孤立性)及多发性两型,其中以单发性远为多见,多发性者为常染色体显性遗传性疾病,多处长骨受累,并伴有骨发育不良及弯曲或短缩畸形。但无论单发或多发性者,其形态学基本相似。
【病变】
本瘤好发于四肢长骨的干骺端,尤以股骨下端、胫骨上端最为多见,其次为肱骨上端,而位于手足骨者极少。
肿瘤大小不等,直径一般为3~4cm,大者可达10cm以上。肿瘤可分为宽基型与带蒂型两种,从骨表面向外隆起,表面呈半球状、菜花状或息肉状。骨软骨瘤结构较特殊,一般可分为三层。①表层为一薄层纤维组织组成,即软骨膜,和相邻骨膜相连;②中层为软骨帽盖由灰白略带蓝色的透明软骨组成,其厚度随患者的年龄而异,年龄越小,软骨帽越厚;在成年人,软骨帽很薄,或几乎消失,其厚度多在1~5mm之间,镜下与正常软骨骺板相似,表层软骨细胞及基质组织较不成熟,愈近底层愈成熟,交界处的成熟软骨细胞排列成柱状,并见钙化及骨化现象。③基底部为肿瘤的主体,常占肿瘤的大部分,由海绵状松质骨组成,骨小梁间多为纤维组织,有较丰富的毛细血管网(图17-3)。基底部下方与正常骨相连。
图17-3 骨软骨瘤
图示特征性的三层结构,即最表浅的纤维层,中间的软骨层(软骨帽)和深部的松质骨
【临床病理联系】
骨软骨瘤一般无明显症状,只是局部肿块形成。有时肿块压迫周围组织,引起疼痛和不适。如果肿块体积迅速增大,软骨帽增厚至1cm以上,则须考虑恶变的可能,这在多发性者比单发性者多见。本瘤如手术切除不彻底,则易复发,多发生在1年或数年后。
(二)软骨肉瘤
软骨肉瘤(chondrosarcoma)是从软骨细胞发生的原发性恶性骨肿瘤,由肿瘤性软骨细胞及软骨基质组成。软骨肉瘤是颇为常见的恶性骨肿瘤,其发病率仅次于骨肉瘤,其发病年龄多在中年以后,多见于40~70岁,根据发病部位不同,可分为中央型及周围型两种。中央型从骨髓腔发生,肿瘤为骨皮质所包绕或穿破骨皮质,多见于长管状骨,特别是股骨和胫骨;周围型从骨肿瘤表层出发,向周围软组织及骨皮质侵犯,多见于骨盆、肩胛骨及肋骨等。少数软骨肉瘤来自软骨瘤和骨软骨瘤之恶变。
【病理变化】
肉眼观,中央型软骨肉瘤主要发生在骨髓腔内,呈灰白色、半透明的分叶状肿物,其中常见淡黄色的钙化或骨化小灶。这些钙化或骨化小灶在X线片上可以看到,对诊断软骨肉瘤很有帮助,但高度恶性的软骨肉瘤钙化常不明显。随肿瘤的增大可使骨髓腔变大并侵犯骨皮质,骨外膜受刺激后,可有反应性新骨形成,使受累骨皮质增厚。恶性程度较高的软骨肉瘤,在早期即可穿破骨皮质,向软组织内扩展,形成较大的肿块,周围没有新骨形成。外围型软骨肉瘤瘤体主要在骨外,其表面被覆一层薄而不完整的包膜。以上两型软骨肉瘤均常发生粘液变、出血及囊性变等继发性变化。
镜下观,肿瘤的分化程度差异很大,分化好的软骨肉瘤在镜下易误诊为软骨瘤,但在肿瘤的边缘可以找到瘤细胞的异型性,如核肥大、深染,出现较多的双核、巨核和多核瘤巨细胞,并可见明显核仁(图17-4)。在分化差的软骨肉瘤则上述瘤细胞的异型性很明显,核分裂像也多见。软骨肉瘤的基质可为与一般透明软骨相似的透明基质,也可为粘液样基质,常见于恶性程度高的软骨肉瘤。
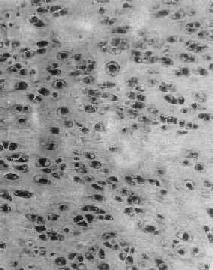
图17-4 软骨肉瘤
软骨细胞大小不等,有的较大。许多细胞有肥硕的胞核,有的有双核
【临床病理联系】
局部疼痛及肿块往往是软骨肉瘤的主要症状。近关节的肿瘤常影响关节活动。盆骨的巨大软骨肉瘤可压迫邻近器官,引起相应症状。软骨肉瘤的分化程度对临床经过有一定影响,分化较好的软骨肉瘤往往生长较慢,预后较好。软骨肉瘤一般比骨肉瘤生长慢,转移也较晚。血道转移可至肺、肝、肾及脑等处,淋巴结转移极罕见。软骨肉瘤术后常易复发,多次复发常使恶性程度增加。
(三)骨巨细胞瘤
骨巨细胞瘤(giant cell tumor of bone)是一种具有局部侵袭性及复发倾向的原发性骨肿瘤。由梭形和卵圆形的基质细胞(stromal cell)及大量散布在其间的多核巨细胞组成,肿瘤间质富于血管。由于本瘤内有大量多核巨细胞,因而称为巨细胞瘤;又因过去认为这些多核巨细胞是破骨细胞,故也称为破骨细胞瘤(osteoclastoma)。本瘤的组织来源未明,以上各名称纯系形态学的描述性命名。在我国骨巨细胞瘤的发病率较高,仅次于骨软骨瘤和骨肉瘤,居第三位。本瘤好发年龄为20~40岁的青壮年,性别无明显差异。
【病理变化】
骨巨细胞瘤多发生于四肢长骨的骨骺端,尤以股骨下端及胫骨上端为多见,约占半数左右,其次为桡骨下端、尺骨下端或肱骨上端等部位。长骨以外则以脊椎为多见。
肉眼观,本瘤常侵犯骨骺线已闭合的长骨端,大多数位于骨骺,早期常为偏心性生长,增大的肿瘤使骨皮质受累而向外膨胀。在肿瘤周围往往有菲薄的骨壳,乃由骨内、外膜反应性新生骨构成,肿瘤的境界比较清楚。肿瘤内原有松质骨大部分或全部消失,瘤内常有纤维组织或骨性间隔。由于肿瘤组织的溶骨性破坏,常造成病理性骨折(图17-5)。肿瘤组织呈灰红色,质软而脆,较大的肿瘤常合并出血及坏死,并伴有囊性变而形成大小不等的空腔,囊腔内含有浆液性或血性液体。晚期病例骨性包壳如果被破坏,则可侵犯软组织形成肿块。关节软骨有抗肿瘤浸润的作用,关节软骨下骨间质可完全被破坏,致使关节软骨失去支持而扭曲变形。
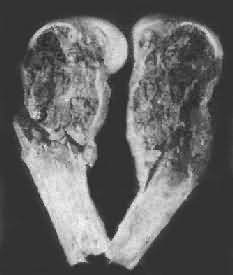
图17-5 骨巨细胞瘤
肿瘤位于肱骨上端,骨皮质破坏,有病理性骨折
镜下,肿瘤主要由单核基质细胞及多核巨细胞等两种细胞组成(图17-6),间质血管丰富。基质细胞为梭形、卵圆形或圆形,细胞境界不清楚,常见胞浆突起。细胞核较大,染色质量中等,可具有一个核仁。多核巨细胞常较均匀地散布在基质细胞之间,是为本瘤的特点。多核巨细胞的直径常为30~60μm,核数一般为15~20个,最多可达100个以上,常聚集在细胞的中央。核的形态与单核基质细胞相似。细胞边界不规则,但分界较清楚,胞浆丰富,略呈嗜碱性,有时还可见含大量脂类的泡沫细胞。本瘤间质血管丰富,有多少不等的胶原纤维。肿瘤本身无成骨现象,但有时见有类骨组织及新生骨小梁,常见于纤维组织的周围,可能是一种反应性新骨形成或病理性骨折后形成的骨痂。
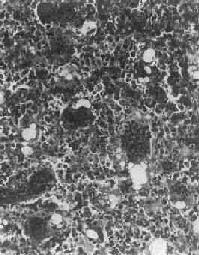
图17-6 骨巨细胞瘤(Ⅰ级)
肿瘤由大量基质细胞和多核巨细胞两种细胞组成
【病理分级】
骨巨细胞瘤在病理组织学上,根据基质细胞和多核巨细胞的数量及异型性的程度分为三级:基质细胞分化好,形态大小一致,偶见核分裂像。多核巨细胞数量多、体积大、细胞核多者为Ⅰ级;基质细胞分化差,异型性明显,细胞密度高,核分裂像多,具有肉瘤样改变。多核巨细胞数量少、体积小、细胞核也少,有明显的异型性者为Ⅲ级;介于两者之间者为Ⅱ级。Ⅰ级基本为良性,具低度侵袭性,刮除术后可复发。多次复发后可变为恶性。Ⅲ级呈恶性肿瘤的表现,易复发和转移至肺。病理分级对判断骨巨细胞瘤的良恶性程度和预后以及治疗方法的选择有一定的参考价值,但并非绝对可靠。
【临床病理联系】
早期症状是局部疼痛及压痛,疼痛性质可为间歇性。位于浅表部位者,可出现局部肿胀或肿块。当肿瘤增大而使表面骨皮质膨胀变薄时,触之有捏乒乓球样感觉。位于脊椎的肿瘤,可引起相应神经压迫症状。
(四)骨肉瘤
骨肉瘤(osteosarcoma)是指肿瘤细胞能直接形成肿瘤性类骨组织或骨组织的恶性肿瘤。骨肉瘤是原发性骨恶性肿瘤中最常见者,约占骨恶性肿瘤的1/3。骨肉瘤的发病率在男性略高,可发生于各级年龄,但最多见于11~20岁,其次为21~30岁,年龄越大,发病率越低。骨肉瘤多发生在骨骼生长发育的旺盛时期,其恶性程度又较高,因此是严重影响劳动生产力并危及生命的重要肿瘤之一,早期诊断及早期治疗具有特别重要意义。
【病因】
骨肉瘤病因不明,其发生与下列因素有关。①骨骼的活跃生长。②放射线:实验证明凡能在骨骼内积存的放射性物质均可诱发骨肉瘤;某些骨疾患如骨巨细胞瘤、动脉瘤性骨囊肿或骨外肿瘤如乳腺瘤、视网膜母细胞瘤等的局部放射线照射治疗,偶尔可引起继发性骨肉瘤。③遗传:视网膜母细胞瘤基因(Rb基因,位于染色体13q14,目前已知它是一种抑癌基因)突变或缺失的遗传性视网膜母细胞瘤患者,发生骨肉瘤的危险性远远高于一般人。近年发现一些骨肉瘤患者也有Rb基因的突变。④病毒:实验证明,动物的骨肉瘤与病毒感染有关,但对人类骨肉瘤尚未有确切的材料说明与病毒的关系。⑤良性骨疾患的恶变:如多发性骨软骨瘤、骨Paget病、骨纤维结构不良等可恶变而发生骨肉瘤,亦称为继发性骨肉瘤。
【病理变化】
骨肉瘤可发生于任何骨,最常见于四肢长骨,半数以上发生于股骨的下端及胫骨或腓骨的上端,其次为肱骨上端。颌骨、脊椎骨、肩胛骨和髂骨等较少见。长骨的骨肉瘤发病年龄较小,发生于扁骨者年龄较大。大多数骨肉瘤发生于骨的内部或中央,在长骨位于干骺端,肿瘤在骨髓腔内及向周围骨皮质浸润形成肿块。因骨骺软骨对骨肉瘤的浸润具有一定的抵抗力,在骨骺板闭合骨化之前(约17~20岁),一般不侵及骨骺端。少数骨肉瘤发生于骨表面,称为皮质旁骨肉瘤,其临床、X线和病理表现均与一般骨肉瘤不同。
肉眼观,长骨肿瘤位于干骺端的骨髓腔中央或为偏心性。一侧或四周的骨皮质被浸润和破坏,其表面的骨外膜常被掀起。在切面上可见肿瘤上、下两端的骨皮质和掀起的骨外膜之间形成三角形隆起,其间堆积由骨外膜产生的新生骨。此三角称为Codman三角,这在X线照片中可以显示出来。在骨外膜被掀起时,自骨外膜通往骨皮质的小血管因受到牵拉而呈垂直于骨皮质分布,在这些垂直的小血管周围,组织的血液供应丰富,故新骨形成增多,这些反应性新生骨小梁呈放射状与骨表面垂直分布(图17-7),在X线上表现为日光放射状阴影,这种现象与上述Codman三角在X线上对骨肉瘤的诊断具有特征性。
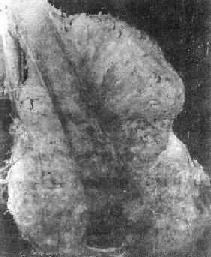
图17-7 硬化性骨肉瘤
股骨干骺端呈梭形肿大,骨干被包埋于中央,周围有清晰的放射状骨梁
骨肉瘤的切面呈多彩状,其外观取决于肿瘤性骨质及软骨的含量以及出血、坏死等继发改变的程度。例如,肿瘤性骨质成分较多时则肉眼观呈黄白色,质地坚硬,有砂粒小点或条纹;如软骨形成明显时则呈半透明状;在肿瘤细胞丰富部位则呈灰红色鱼肉状。
镜下,骨肉瘤由明显间变的梭形或多边形肉瘤细胞组成,细胞大小不等,核形奇异,大而深染,核仁明显,易见病理性核分裂像。肿瘤细胞直接形成肿瘤性类骨组织或骨组织,是诊断骨肉瘤的最重要的组织学依据。所形成的类骨组织或骨组织在不同肿瘤或同一肿瘤的不同部位多少不等。往往可看到肿瘤性骨质发生过程中各阶段的形态,最早期在恶性肿瘤细胞间出现均质红染的胶原样物质,其后红染物质逐渐增多,将肿瘤细胞分隔疏远,构成小梁或片状的肿瘤性类骨组织(图17-8)。类骨组织可伴钙盐沉着,其内的肿瘤细胞固缩变小,形成肿瘤性骨质。骨肉瘤内也可出现肿瘤性软骨(图17-9)。
图17-8 骨肉瘤
多形性肉瘤细胞直接形成肿瘤性骨样组织
图17-9 骨肉瘤
骨肉瘤内肿瘤性软骨
骨肉瘤起源于间叶细胞,形态上表现出多方向分化的潜能,而使骨肉瘤的成分较为复杂。根据其主要的分化成分,组织学上可分成几种类型:最常见者为骨母细胞型,以异型骨母细胞为主要成分,细胞呈多边形,较肥硕,核圆、核仁明显,胞浆丰富,伴较多的肿瘤性类骨和骨组织形成;由软骨肉瘤样组织为主要成分者,属软骨母细胞型;以梭形细胞和胶原纤维为主,似纤维肉瘤者为纤维母细胞型;肿瘤内有许多显著扩张的血管腔隙,伴较多破骨细胞型多核巨细胞,甚似动脉瘤性骨囊肿者为血管扩张型,此型十分少见。在同一肿瘤中上述各种类型的成分常混合存在,但以某一类型为主。不论何种类型,都能见到肿瘤细胞直接形成肿瘤性类骨和骨组织,这是区别于其他骨肿瘤的特征。
【临床病理联系】
骨肉瘤最早的症状是局部疼痛,日渐加剧,持续不断,以夜间为明显。发病2~3月后,局部出现肿胀、质地坚硬。患部皮肤紧张,多呈紫铜色,表面静脉曲张。如骨皮质受侵犯时,轻度外伤即可致病理性骨折。
血清碱性磷酸酶的增高是骨肉瘤的唯一重要化验室检查指征,这和瘤组织内碱性磷酸酶的含量增高相一致,约见于半数病例,对骨肉瘤诊断和推测预后有一定价值。
X线检查对骨肉瘤的诊断有重要价值。常见到肿瘤性骨质形成,表现为云絮状或斑块状密度增高的阴影。这类病例在X线诊断时称为硬化型骨肉瘤。少数病例呈溶骨型,病理上主要为软骨母细胞型和纤维母细胞型以骨质破坏为主,呈虫蚀状或大片状骨质破坏,边界模糊。如前所述,X线检查如发现Codman三角或日光放射状阴影,则对骨肉瘤的诊断很有帮助。
骨肉瘤是高度恶性肿瘤,一般生长迅速,预后较差。局部扩展一方面侵犯骨髓腔及骨皮质,并破坏骨膜及周围软组织;另一方面向骨骺蔓延,甚至扩展到关节软骨。极少数病例可越过关节软骨,侵入关节囊,造成关节活动障碍。近年还发现,骨肉瘤在骨内可呈跳跃性转移(skip metastasis),即在与原发瘤同一骨内的另一处形成孤立性转移结节,甚至还可以转移到相邻的骨内,形成孤立结节,约见于1/4的病例,且不易被X线检查发现,因而预后更差。远处转移多经血道到肺,不少病例在发现原发瘤的同时,即已有肺转移。极少数病例可转移到局部淋巴结。
皮质旁骨肉瘤是骨肉瘤的一种特殊亚型。与一般骨肉瘤不同,本瘤发生于骨膜或骨旁的成骨性纤维组织,瘤体的主要部分位于骨的表面。大多生长缓慢,恶性程度较低,预后远较一般骨肉瘤好,其发病年龄比一般骨肉瘤大5~10岁。本瘤好发于股骨下端的后方,其次为股骨上部。瘤体一般较大,边界较清晰,可呈分叶状。早期对骨质侵犯较少,故易于剥离;晚期可破坏骨皮质而侵犯骨髓腔,但远处转移少见。在切面上,靠近骨皮质或骨膜部分质坚实,而瘤体边缘部分较软,为富于瘤细胞部分。瘤体一般不见出血及坏死。镜下,为分化好的骨肉瘤,成骨现象比较明显。在肿瘤性骨小梁间为增生较活跃的纤维组织,间变不明显,形态上似分化好的纤维肉瘤。少数皮质骨肉瘤分化较差,形成骨质较少,与骨内骨肉瘤相似。
第三节 佝偻病和骨软化症
佝偻病(rickets)和骨软化症(osteomalacia)是以骨基质钙盐沉着障碍为主的慢性全身性疾病,表现为骨组织内类骨组织(未钙化骨基质)的过多聚积。病变如发生在生长中的骨骼,则成佝偻病,多见于婴幼儿,称为婴幼儿佝偻病。发生在年龄较大的儿童者,称为晚期佝偻病,较为少见。病变如发生在成年人,骨的生长已停止者,则称为骨软化症。佝偻病和骨软化症在病因及病变方面基本相同。
【病因和发病机制】
佝偻病和骨软化症主要是维生素D缺乏,钙磷代谢障碍,类骨组织钙化不良所造成的骨骼病变。
维生素D为脂溶性类固醇衍生物,以维生素D2(麦角钙化醇)尤其以维生素D3(胆钙化醇)较为重要,两者代谢和功能类同。维生素D可来自食物,维生素D2的前身物质麦角固醇存在于植物和谷物中,在体内外经紫外线照射后可转化为人体能吸收的维生素D2;维生素D3主要存在于肝、奶、和蛋黄中,以鱼肝油含量最多。食物来源的维生素D在小肠以乳糜微粒形式吸收,与胆汁酸盐的作用有关。维生素D的另一重要来源为体内合成,日光中紫外线使表皮基底细胞内的维生素D2前身麦角固醇和大量存在的维生素D3前身7-脱氢胆固醇(在体内由胆固醇脱氢而成)转化为维生素D2和维生素D3。维生素D2和D3为非活性物质,可与血浆中的α-1球蛋白结合储存于脂肪组织中。血液中维生素D2和D3经肝细胞线粒体中25-羟化酶的作用,转化为25-羟维生素D2(25-OHD2)和25-羟维生素D3(25-OHD3)后才具有活性。以后又经肾小管上皮α-1羟化酶进一步羟化为1,25-二羟维生素D2(1,25-(OH)2D2)和1,25-二羟维生素D3(1,25-(OH)2D3),活性大大提高,而有类似于激素的作用。1,25-(OH)2D受其自身合成的反馈调节,血浆中1,25-(OH)2D浓度升高可抑制肝内25-羟化酶的作用,使25-OHD和1,25-(OH)2D形成减少;反之,则增加。它还受血清钙、磷浓度和甲状旁腺激素以及降钙素的调节。低血钙时,甲状旁腺激素分泌增加,使肾α-1羟化酶活性增高,1,25-(OH)2D合成增加;高血钙时则相反。低血磷时亦可增加1,25(OH)2D的合成。
维生素D通过对小肠、肾和骨三个靶器官的作用,维持和调节血浆钙和磷的水平,它对骺板软骨和类骨组织的钙化是必不可少的。①1,25-(OH)2D明确地促进小肠粘膜上皮对钙磷的吸收。目前认为是通过受体-基因介导作用(receptor-gene mediated function)实现的,1,25-(OH)2D与受体结合形成的复合物促进mRNA转录和合成特异的钙磷输送蛋白,从而增加钙磷的吸收。②1,25-(OH02D可促进肾近曲小管对钙磷的重吸收,但其作用较弱且尚不完全明确,需与甲状旁腺激素协同作用。③1,25-(OH)2D对骨有二种相反的作用。一方面,骨是人体的钙库,当血钙降低时,1,25-(OH)2D与甲状旁腺激素协同作用,从骨回吸收钙磷,维持血浆钙磷的正常浓度。其机制是很可能通过破骨细胞的作用,使骨盐溶解。亦有证据表明,它们可能通过抑制骨母细胞合成Ⅰ型胶原,使骨重建暂停,骨盐被回吸收。另一方面,1,25-(OH)2D促进骺板软骨和类骨组织的钙化,这与它和甲状旁腺激素维持钙、磷在血浆中的饱和状态有关,有利于骨盐的沉积。亦有人认为1,25-(OH)2D可增加类骨组织中钙结合蛋白,即骨钙蛋白(osteocalcin)和骨联蛋白(osteonectin)的合成有关,它们使类骨周围组织液中的钙和磷与类骨中的Ⅰ型胶原分子结合,类骨因此而钙化。
维生素D缺乏性佝偻病或骨软化病多见于骨骼生长快、对维生素D需要量高的婴幼儿和妊娠妇女或多产妇。维生素D摄入不足,尤其是日光照射不足是重要的原因,因占人体需要量80%的维生素D可来自体内合成。此外,胃肠以及胆道疾患,影响脂溶性维生素D和钙磷的吸收,也是原因之一。
维生素D缺乏,主要是1,25-(OH)2D3缺乏时,钙磷从肠道吸收减少,导致血钙磷下降,而促使甲状旁腺激素分泌增加,从而骨质脱钙,使血钙维持正常,但肾小管对磷的重吸收减少,使尿磷增加而血磷减少。这样,维生素D缺乏时,血钙在正常或偏低水平,而血磷减少。结果使钙磷浓度积(正常在34~40)降低,钙磷不能在骨基质中充分沉积,导致类骨组织大量堆积,造成佝偻病或骨软化病。
严重的肝损害时,肝细胞25-羟化酶合成障碍,维生素D羟化不足,而引起肝性佝偻病。慢性肾功能不全时,肾小管合成α-1羟化酶不足,而使1,25-(OH)2D减少,钙磷吸收减少,又因肾小球滤过功能下降,磷酸盐潴留,以致血磷偏高,血钙则减少。另外,肾小管分泌H+和重碳酸盐重吸收障碍,亦使钙排出增加。上述改变促使甲状旁腺分泌增加,骨质脱钙,钙盐沉积障碍,而引起肾性佝偻病。
【病理变化】
1.佝偻病病理变化 佝偻病时由于膜内化骨及软骨化骨的钙化过程均发生障碍,因此长骨和扁骨均同样受累。
(1)四肢长管状骨:骺板软骨、干骺端及骨干均可不同程度受累。骺板软骨是骨生长最活跃的部位,正常时软骨内化骨必须通过软骨细胞增生区内软骨细胞和基质不断退化和钙化以及不断被破骨细胞清除、吸收,同时血管和骨母细胞侵入形成类骨组织,进而钙化成骨组织。佝偻病时,软骨细胞增生区钙化、吸收受阻,软骨组织大量堆积并突向干骺端侧,呈半岛样或舌状生长(图17-10)。同时软骨区内所形成的类骨组织也不能钙化或钙化明显不足,从而构成软骨组织和干骺端类骨组织相互混杂的中间带,致使在正常状态下本应呈一条整齐面狭窄的骨骺线显著增宽,而且变得参差不齐,在X线片上构成骺板软骨带明显增宽,钙化带模糊不清呈毛刷状。此外干骺端下的骨膜内化骨也有钙化障碍及类骨组织堆积,使干骺端膨大增宽,X线片上呈杯口状改变。骨干的骨膜内化骨同样也有钙化障碍,因此骨皮质表面和骨皮质的近髓腔侧,都有大量类骨组织堆积,使骨髓腔变窄,长骨横径增加。由于骨质缺钙,类骨组织缺乏承受力,在重力作用下长骨骨干可变弯曲,尤以胫骨和股骨最易变形,形成X形腿或O形腿。以O形腿为多见。
图17-10
骨骺线不整齐、变宽、软骨呈舌状向骨干伸展
(2)颅骨及肋骨:在婴幼儿颅骨的病变常很明显,常在佝偻病的早期即可出现。颅骨骨缝及囟门闭合常延迟或不完全,因此头形常较大,囟门部呈结缔组织性膜样结构。此外,由于额骨前面的两个骨化中心和顶骨的两个骨化中心都在膜内骨化过程中发生钙化障碍,因此类骨组织在颅骨的四角堆积并向表面隆起,形成方形颅。颅骨由于骨化停止,严重者骨质菲薄,批压时凹陷,并有如乒乓球样的弹性感。
肋骨和肋软骨结合处的改变与长骨骺板及干骺端的改变相似,由于软骨及骨样组织的堆积,致使肋骨和肋软骨的结合部呈结节状隆起。因多个肋骨同时受累,故结节状隆起排列成行,形似串珠,称为佝偻病串珠(rachitic rosary)(图17-11),常是佝偻病的较早期表现之一。此外,肋骨因含钙量少,缺乏韧性,同时由于膈在呼吸时的长期牵拉,在胸壁前部左右两侧各形成横行的沟形凹陷,称为Harrison沟。又因在呼吸时,肋骨受肋间肌的牵拉而下陷,使胸骨相对向前突出,形成鸡胸畸形。
图17-11 佝偻病的肋骨
肋骨和肋软骨结合部呈结节状隆起,排列成行,形似串珠
除上述常见的佝偻病改变外,还有两种较少见的佝偻病,即①先天性或胎儿性佝偻病,在婴儿出生时已有佝偻病表现,主要是由于母亲在怀孕时有严重的维生素缺乏所致。②晚期佝偻病,多见于北方地区,发病多在10岁以后的儿童,故其改变介乎婴幼儿佝偻病和骨软化症之间。因此时颅骨的骨化已基本完成,而肋骨生长较慢,故方形颅和肋骨串珠等均不显著。骨骼生长较慢,严重时可形成侏儒畸形。
2.骨软化症病理变化骨软化症发生于成人,其改变与佝偻病相似。因成人的骨发育已停止,故其改变限于膜性化骨的钙化障碍,致过量的类骨组织堆积在骨的表面,骨质变软,同时因为承重力减弱而导致各种畸形,常见的有骨盆畸形,脊柱侧突及长骨弯曲等。骨盆畸形表现为骨盆的前后径及左右径均变短,耻骨联合处变尖而向前突出,呈鸟喙状,称为喙状骨盆。
附 骨质疏松症
骨质疏松症(osteoporosis)是指骨小梁的数量绝对值减少,而骨小梁的结构及骨基质的钙化均正常,因此与骨软化不同,故也有人称之为骨质缺乏症(osteopenia)。骨质疏松乃由于各种原因引起的骨形成减少或骨吸收增强或两者兼而有之所致。骨质疏松症可分为局限型与弥漫型两种。
1.局限型骨质疏松症(或废用性骨质疏松症) 多因患肢的长期不活动或瘫痪引起,如见于小儿麻痹症或骨结核治疗时,大约数周内即可出现,表现为松质骨的小梁减少、变细,皮质骨变薄、变疏松。X线检查可以早期发现,病变为灶性,特别在软骨下的关节或干骺端明显,可能因该处骨质代谢较旺盛所致。在肌肉恢复运动时,骨小梁形成增加,可逐渐恢复正常状态,尤其在小儿比较明显。
2.全身性骨质疏松症由于:①营养缺乏:如蛋白质、钙或维生素C或D缺乏。②多种内分泌系统疾患,如Cushing综合征、甲状腺功能亢进或性腺功能低下。以上也称为继发性骨质疏松症。另有一种原发性或特发性骨质疏松症(primary or idiopathic osteoporosis),常见于中年以后,女性比男性多见,特别是绝经期后妇女更为多见,故也称为老年或绝经期后骨质疏松症(senile or post menopausal osteoporosis)。其发生机制尚未完全阐明,可能由于雌激素缺乏,促进骨质吸收有关。患者血清碱性磷酸酶及钙、磷水平均正常,故和骨软化不同。有人报告服用雌激素可使此种骨质吸收停止,并增加钙的存留。本病也可见于卵巢切除后的年轻妇女。
全身性骨质疏松症可导致脊柱塌陷,椎骨凹陷,常易发生压迫性骨折及疼痛。长骨的轻微外伤即可引起骨折,尤其常见于股骨颈部,其次为腕及肱骨上端。
第四节 类风湿性关节炎
类风湿性关节炎(rheumatoid arthritis)是一种慢性全身性自身免疫性疾病。主要侵犯全身各处关节,呈多发性和对称性慢性增生性滑膜炎,由此引起关节软骨和关节囊的破坏,最后导致关节强直畸形。除关节外,身体其他器官或组织也可受累,包括皮下组织、心、血管、肺、脾、淋巴结、眼和浆膜等处。本病发病年龄多在25~55岁之间,也见于儿童。女性发病率比男性高2~3倍。本病呈慢性经过,病变增剧和缓解反复交替进行。绝大多数患者血浆中有类风湿因子(rheumatoid factor,RF)及其免疫复合物存在。
【病理变化】
(一)基本病变
类风湿性关节炎作为一种全身性免疫性疾病,在关节和其他受累器官及组织内,有与免疫反应密切相关的淋巴细胞、浆细胞和巨噬细胞浸润,并可伴淋巴滤泡形成。另外,本病病变主要累及结缔组织,属于胶原疾病(collagen disease),全身间质胶原纤维和血管可呈现纤维素样变性或坏死(很可能由局部免疫复合物沉积所致),表现为:①类风湿性肉芽肿(rheumatoid granuloma)或称类风湿小结(rheumatoid nodule)形成,具有一定特征性。镜下,小结中央为大片纤维素样坏死,周围有核呈栅状或放射状排列的类上皮细胞,再外围为增生的毛细血管及纤维母细胞,伴上述炎症细胞浸润。最后则纤维化。类风湿小结主要发生于皮肤、其次为心、肺、脾和浆膜等处。②血管炎:主要发生于小静脉和小动脉,轻重不一,少数严重者出现纤维素样坏死性动脉炎,常伴有血栓形成。
(二)各器官的病变
1.关节病变最常见,多为多发性及对称性,常累及手足小关节,尤其是近侧指间关节、掌指关节及蹠趾关节,其次为膝、踝、腕、肘、髋及脊椎等关节。
(1)滑膜病变:早期,主要病变在滑膜,可分为急性及慢性两阶段,两者间没有明显界限。
急性滑膜炎时关节肿胀,滑膜充血、水肿,表面滑膜组织可见灶性坏死和纤维素被覆。此期虽可见中性粒细胞浸润,但以淋巴细胞和巨噬细胞为主。关节腔内有混浊的乳状积液,或可见纤维蛋白凝块。
慢性滑膜炎具有较特征性的改变,表现为:①滑膜内有大量淋巴细胞、巨噬细胞和浆细胞浸润,并可形成淋巴小结,病程较久者可见生发中心。②滑膜细胞活跃增生,可形成多层状,并可见多核巨细胞。后者胞浆略嗜碱性,核有2~12个不等,多位于胞浆外围呈花环状排列。电镜下,增生的滑膜细胞以B型(纤维母细胞样细胞)为主,而多核巨细胞则形态上与A型滑膜细胞(巨噬细胞样细胞)相似。③滑膜绒毛状增生及血管翳形成。滑膜的慢性炎症,导致新生血管和纤维组织增生,滑膜呈不规则增厚,并形成许多绒毛状突起伸向关节腔。绒毛直径约1~2mm,长度可达2cm。上述淋巴小结常位于绒毛末端。滑膜内可见血管炎改变,或有灶性坏死,或小灶性出血和含铁血黄素沉着,滑膜和绒毛表面可见纤维素沉着。④滑膜内炎性肉芽组织向关节软骨边缘部扩展,形成血管翳(pannus),并逐渐覆盖和破坏关节软骨。
(2)关节软骨变化:急性滑膜炎可以消退而不累及关节软骨,但当炎症反复发作并转变为慢性时,关节软骨几乎必然受损。最早表现为基质的异染性减弱或消失,用甲苯胺蓝染色可以证实。关节软骨边缘形成的血管翳直接侵蚀破坏关节软骨,两者交界面可见软骨糜烂和小灶性坏死。随着血管翳逐渐向心性伸展和覆盖整个关节软骨表面,关节软骨严重破坏,最终被血管翳取代。
长期的慢性炎症和反复发作,滑膜不断增生,纤维组织日益堆积,关节腔内纤维素性渗出物又不断机化和瘢痕化,使关节腔变窄,同时关节软骨破坏和被血管翳取代,两关节面发生纤维性粘连,形成纤维性关节强直,最后可发展为骨性关节强直。由于关节周围肌肉痉挛及肌腱松弛,可造成关节脱位或半脱位,加重了关节畸形。
(3)关节相邻组织的变化:①慢性类风湿性关节炎会引起关节邻近骨组织吸收和骨质疏松以及关节软骨下骨质破坏,有时可见小囊腔形成,偶尔附近骨皮质被侵蚀破坏,可导致病理性骨折。这些改变与破骨细胞和巨噬细胞进行骨质吸收、长期应用皮质激素类药物治疗以及受关节炎症波及等有关。②关节附近肌腱、韧带和肌肉常受累,有局灶性淋巴细胞、浆细胞和巨噬细胞浸润,偶见类风湿小结。肌肉有废用性萎缩。③关节病变的引流淋巴结肿大,淋巴组织增生,生发中心明显,偶见类风湿肉芽肿形成。
2.关节以外的类风湿病改变并不常见,多伴发于有明显活动性关节病变者。
(1)皮下结节:是关节以外类风湿病中最常见者,见于约20%~25%的病例,多位于关节旁,最常于鹰嘴突等骨质突出和受压部位。单个或多个,大小由数毫米至2cm不等,质硬、无压痛。肉眼观呈灰白色,中央为黄色坏死区,镜下呈典型类风湿性肉芽肿改变。皮下结节存在的时间较长,可持续数月或数年不退。
(2)心和肺等病变:类风湿性肉芽肿、血管炎和淋巴细胞、浆细胞和巨噬细胞浸润等改变可出现于许多器官和组织,但较常见于心脏(心内膜、心肌和心外膜)和肺,最终导致心和肺灶性或弥漫性间质纤维化。偶尔引起心瓣膜变形和关闭不全。浆膜受累造成纤维素性心包炎和胸膜炎,最后引起心包和胸膜广泛增厚、粘连。
(3)血管病变:偶尔出现急性纤维素样坏死性动脉炎,常伴血栓形成和引起相应组织的梗死。主动脉亦可受累。
【病因和发病机制】
目前多认为本病属于一种自身免疫性疾病,其始动因子尚不清楚,可能是感染因子(如病毒、支原体或细菌等)进入人体后,其所含某些成分(如寡糖或糖肽碎片)被关节内滑膜细胞摄取并组合到滑膜细胞所合成的蛋白多糖中,使其结构发生改变而具抗原性。这种自身抗原不仅可使机体产生抗体(IgG),同时还导致IgG分子的Fc片段结构发生改变,形成新的抗原决定簇,从而激发另一种抗体形成,即类风湿因子(RF)。血清中RF最主要的成分是IgM,亦有IgG、IgA和IgE等。IgM型的RF约见于85%~95%的类风湿性关节炎患者,是临床诊断的重要指标。各种免疫球蛋白类型的RF与IgG形成的免疫复合物存在于血循环中。RF和免疫球蛋白可以在关节内合成并结合成免疫复合物,循环中RF-IgG复合物亦可以沉积于局部组织,这与关节和关节外器官和组织病变的发生有密切关系。关节滑膜内RF-IgG复合物可以固定及激活补体,产生C3a和C5a,吸引中性粒细胞和单核细胞渗出。中性粒细胞、单核细胞及滑膜细胞(A型细胞)吞噬了上述免疫复合物后,被激活并合成和释放溶酶体酶,包括中性蛋白酶、胶原酶等以及各种介质,如前列腺素、白三烯、IL-1等,导致滑膜及关节软骨的破坏。IL-1是类风湿关节炎的主要介质,由激活的巨噬细胞和滑膜细胞产生。IL-1可使滑膜细胞和软骨细胞合成和释放胶原酶和其他蛋白溶解酶,并抑制软骨细胞合成蛋白多糖,本身又是一种破骨细胞激活因子。滑膜内不仅有RF、各种免疫球蛋白及补体等,而且经免疫荧光和组织培养亦说明它们可由滑膜内B细胞和浆细胞产生。即使在始动因子(如感染因子)已不复存在的情况下,RF仍不断产生,结果导致炎症病变反复发作,成为慢性炎症。
研究结果表明,除上述体液免疫因素外,本病与细胞免疫亦有密切关系。随滑膜病变转为慢性,T细胞和浆细胞明显增加,其中主要是T4辅助细胞。T4与B细胞协同作用,参与RF和免疫球蛋白合成,滑膜内HLA-DR阳性巨噬细胞和树突细胞增加,与T4相互作用,亦与造成关节损害的免疫机制有关。
关于感染因子与本病的关系,近年来注意到EB病毒感染的作用。约65%~93%的类风湿性关节炎患者血清中有EB病毒核心抗体,而其他关节炎患者则仅为10%~29%;又本病患者细胞培养的B细胞,经EB病毒转化后可产生RF。
第五节 大骨节病
大骨节病是以关节软骨变性、坏死为主的慢性地方性疾病。疾病晚期,继关节软骨变性、坏死而出现关节周围代偿性软骨及骨质增生,使关节周径显着增粗变形,故我国东北病区称之为大骨节病。又因本病造成患者行走困难,故我国西北一带又俗称为柳拐子病。国际上通称为Kashin-Beck病。
本病多发生在山区潮湿寒冷地区,有明显的地区分布,患者离开病区后病情多停止发展。在我国陕西、山西、甘肃、内蒙、河南、黑龙江、辽宁及吉林等省流行甚广。在国外,前苏联、朝鲜、日本及瑞典亦有本病发生。
大骨节病多自童年发病,呈慢性经过,在青春期前后症状变明显。早期症状是四肢麻木、关节疼痛、运动不灵活,关节附近肌肉挛缩。以后,关节肿大变形,运动受限制,伴有疼痛及摩擦音。病变多自手指、足趾等管状骨开始,以后侵犯膝、肘、肩及股等较大关节,尤以负重较大或运动较强的关节病变最明显。如果在青少年时期发病而且较为严重时,可因骺板软骨被侵犯,早期化骨,骨生长早期停止,而引起短指和短趾畸形(图17-12);发病越早,病变越重,长骨的生长发育也越差,患者成年后身材矮小(大骨节病性侏儒)。
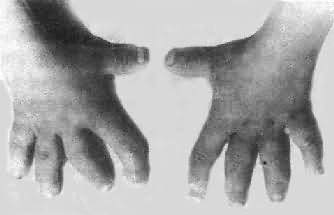
图17-12 大骨节病
双手呈短指畸形
【病理变化】
除胸锁关节外,全身关节均有不同程度的病理变化,但主要病变在四肢管状骨的关节软骨及骺板,下肢较上肢为严重。病变呈对称性,左右两侧关节同时受累,主要以软骨的变性及坏死为特征。
1.关节软骨病变最早的改变是软骨基质的变性。基质由正常轻度嗜碱性变为嗜酸性,呈深红染。进而基质开始溶解、变稀薄淡染,以致显现排列方向较一致的胶原纤维(原纤维显现)。病变如进一步发展则液化溶解形成空泡或裂隙。随着基质的变性,软骨细胞发生空泡变性,严重时,胞浆变为蜂窝状,胞核常被挤到一侧。有时可见软骨基质中水分增多,液化而淡染,其中游离的软骨细胞由正常的圆形变为星芒状,似粘液细胞,称为软骨粘液变性。变性严重时可发生坏死,软骨细胞核固缩消失,最后只剩下细胞轮廓。
软骨的坏死首先出现于关节软骨的深层与骨质交界处,早期为灶性或片状,以后相邻病灶互相融合而形成明显的坏死带(图17-13),有人认为这种带状坏死对本病有诊断意义。
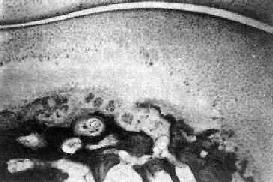
图17-13 大骨节病
关节软骨深层的带状坏死
在软骨的坏死区边缘或带状坏死的一侧健存的软骨细胞活跃增生,通过细胞直接分裂而形成大小不等的软骨细胞团(图17-14),一般呈圆形或椭圆形,由数个或数十个细胞构成。这些软骨细胞体积较小,其间的软骨基质也很少。
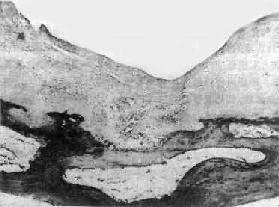
图17-14
关节软骨坏死,其邻近区软骨细胞巢状增生(切片中央部分),软骨下骨板中央部分凹陷
2.骺板软骨及干骺端病变在未成年患者,骺板软骨的病变较明显。骺板软骨可呈现与关节软骨完全相同的变性和坏死。病变轻者,软骨细胞柱变短,排列紊乱,病变重者软骨组织可出现变性及坏死,以后出现不规则早期钙化。骺板软骨下小血管侵入变性和坏死的骺板内,并由破骨细胞溶解、吸收以及形成不规则新生骨组织,造成早期骨化。由于骨化过程紊乱,新生骨小梁不仅较为稀疏变短粗,而且新生骨小梁的方向由正常时的纵向排列变为横向,这是最主要的改变,使骨的纵向生长受阻。横行的骨小梁贴附于骺板软骨表明骨的生长已停顿。
干骺端的松质骨,由于骨小梁稀疏,骨髓腔明显扩大,形成典型的骨质疏松。有时可见骨小梁的断裂或破碎,甚至有小囊腔形成。
大骨节病的肉眼改变,早期在关节软骨不明显。病变较重时关节软骨质地变脆,色暗,表面粗糙,有小沟纹,坏死软骨作瓣状脱落而形成大小不等的溃疡。软骨组织成片脱落时则形成关节内游离体。软骨坏死周边部分及骨骺侧壁的软骨、软骨外膜及肌腱附着部的纤维软骨以及关节软骨周围的滑膜及结缔组织亦常增生,并渐渐骨化,结果在关节周围形成许多骨赘(图17-15);在关节运动的压力作用下,骨赘成为盘状或唇状突起,致使关节肿大,形成大骨节病的外观。增生的滑膜绒毛以及骨赘亦可脱落成为游离体,存在于关节腔内。

图17-15 大骨节病
肱骨、桡骨及尺骨的关节面极不平滑,周边有增生的骨赘
骺板软骨在早期也无明显的肉眼观改变,病变较重时肉眼上骺板厚薄不均,弯曲呈波浪状,与同龄健康人相比,骺板较薄。
【病因】
大骨节病的病因,至今尚不清楚。基本有以下两种观点。
1.病区的粮食及饮水中毒病区中的粮食如小麦和玉米受镰刀菌污染。镰刀菌本身的毒素及被污染谷物所产生的分解产物胺类可引起骨及软骨病变。通过调查发现我国东北及西北病区的粮食常有镰刀菌污染,但两地区污染镰刀菌的种属有所不同。前者为尖孢子镰刀菌,后者为串珠镰刀菌。所谓饮水中毒是指病区饮水中的腐殖质酸(-OH)含量过高,较非病区高6~8倍。腐殖质酸可引起硫酸软骨素的代谢障碍,导致软骨改变。
2.病区土壤、饮水及粮食等的常量化学元素与微量化学元素的比值失调近年来,对于微量元素硒在大骨节病发病中的作用予以极大的关注。调查发现,病区的水、土壤及粮食均呈低硒状态;儿童的头发及尿中硒含量明显低于非病区;应用亚硒酸钠防治大骨节病取得了较好的效果。实验证明,缺硒可影响软骨细胞生物膜的完整性及稳定性,使之容易受有害因子的损伤。硒还可促进硫的代谢和利用,对软骨基质也有一定的保护作用。
总之,病因尚未清楚,低硒及粮食和饮水中毒以及营养缺乏等可能对大骨节病的发病有重要作用。
第十八章 传染病
传染病是由病原微生物侵入人体所引起的一类疾病,能在人群中引起局部的或广泛的流行。传染病的病理过程决定于病原微生物的性质和机体的反应,以及是否及时适当的治疗。
引起传染病的病原微生物有病毒、立克次体、支原体、细菌、真菌、寄生虫等。这些病原体的生物学特性不同,引起病变的机制不同,侵袭的器官也不同。病原体侵入机体的途径不同,如经皮肤、粘膜或由血液扩散至体内。有的病原体长期潜伏,有的进入体内即生长繁殖,产生对机体有害物质,影响机体局部或全身的功能和形态变化,引起疾病。
机体对入侵的病原体多有积极的反应。皮肤、粘膜对于病原体是第一道屏障,不但具有机械的隔离阻挡作用,尚有其它因素作用于病原体,如局部pH,分泌物的各种酶和分解产物,均不利于病原体的繁殖和侵袭。病原体进入体内后,机体的免疫应答系统即积极反应,调动细胞免疫和体液免疫,同时引起局部或全身的炎症反应。不同病原体引起病理改变的基本性质属于炎症范畴。因此传染病的局部和全身反应的变化规律和炎症的规律基本相同。随着病变的发展,临床也出现潜伏期、前驱期、发病期和愈复期。病变发展过程和病程及病原体的数量、毒力以及机体免疫功能和药物治疗有明显的关系。机体抵抗力强和治疗得当可以缩短病程和减轻损伤,否则病原体繁殖、播散,可造成严重的后果。
病原体引起细胞病变的机制不同,其方式有三:①病原体进入细胞内,直接引起细胞死亡;②病原体释放内毒素或外毒素杀伤细胞,或释放酶降解组织成分,或损伤血管引起缺血性坏死;③病原体引起机体免疫反应,进而由于免疫介导机制引起组织损伤。例如,病毒的致病机制是:藉其表面蛋白和机体细胞特种蛋白(受体)相结合而进入细胞,如EB病毒可连结在吞噬细胞的CR2蛋白上而进入细胞。进入细胞后,病毒的核酸(DNA或RNA)进行复制,影响宿主的核酸代谢,抑制其DNA、RNA和蛋白合成;病毒蛋白部分插入宿主细胞的质膜,引起直接损伤;病毒蛋白裸露在宿主细胞表面,引起机体免疫系统和淋巴细胞的攻击;病毒损伤宿主抗微生物能力,引起继发感染。细菌引起细胞病变乃依赖其粘附于宿主细胞和产生毒素。细菌毒素可引起全身性反应,如发热、白细胞增多、休克和巨噬细胞反应等免疫反应。同时肝、脾、淋巴结肿大,以及实质器官如心、肝、肾和神经系统的变性、坏死等。局部器官的病变和病原体种类、器官选择性及其毒素性质有关。
大多数传染病通过机体抵抗力增强和适当的治疗可获痊愈。如抵抗力差而又未获及时治疗,可以转为慢性或蔓延扩散,甚至引起死亡。
单凭病理形态学变化,常常不能对传染病作出及时的和特异的诊断,这是由于早期病变多系一种非特异炎症改变。因此必须结合病原的鉴定方能确诊。病原微生物的检查有病原体形态、病原体分离培养、代谢产物生化特性测定和免疫学方法等。这些方法常受标本取材、病程和治疗的影响,此外尚有操作繁复、费时较长的缺点。
90年代,分子生物学理论和方法引进病理学,形成了分子病理学。DNA探针技术已广泛应用于病理学的研究和诊断,用以诊断传染病可获得快速准确的结果。该方法乃采用病原体单股DNA小片段,以同位素、免疫酶标或荧光素标记,可以检出待查病原体相应的DNA(或基因),从而确诊。它有特异性强,灵敏度高,标本易取,操作简便、快捷等优点。方法学有液相分子杂交和原位杂交等,尤其对检测极微量病原体DNA片段,可应用聚合酶链反应(PCR),使其扩增几十万倍,使之易于检出。本章所叙述的几种传染病几乎全部可以用分子探针进行病原鉴定。
第一节 结核病
结核病(tuberculosis)是由结核杆菌引起的一种慢性传染病。全身各器官均可发生,但以肺结核最为多见。其病变特征是结核结节形成并伴有不同程度的干酪样坏死。
【病因和发病机制】
结核病的病原菌是结核杆菌,对人致病的主要类型为人型和牛型。结核杆菌含有脂质、蛋白和多糖类三种成分:①脂质:特别是脂质中的糖脂更为重要。糖脂的衍生物之一称索状因子(cord factor),能使结核杆菌在培养基上生长时呈蜿蜒索状排列。这种形式生长的结核杆菌在动物体内具有毒力。另一种糖脂为蜡质D(wax D),将其与结核菌体蛋白一起注入动物体内,能引起强烈的变态反应,造成机体的损伤。此外,磷脂还能使炎症灶中的巨噬细胞转变为类上皮细胞,从而形成结核结节。脂质除可能与毒力有关外,还可保护菌体不易被巨噬细胞消化。②蛋白:具有抗原性,与蜡质D结合后能使机体发生变态反应,引起组织坏死和全身中毒症状,并在形成结核结节中发挥一定的作用。③多糖类:可引起局部中性粒细胞浸润,并可作为半抗原参与免疫反应。
结核病主要经呼吸道传染。肺结核(主要是空洞型肺结核)病人在谈话、咳嗽和喷嚏时,从呼吸道排出大量带菌微滴(每个微滴可含10~20个细菌)。吸进这些带菌的微滴即可造成感染。少数病人可因食入带菌的食物经消化道感染。细菌经皮肤伤口感染者极少见。
结核病的发生和发展取决于很多因素,其中最重要的是感染的菌量及其毒力的大小和机体的反应性(免疫反应或变态反应)。后者在结核病的发病学上起着特别重要的作用。
目前一般认为,结核病的免疫反应以细胞免疫为主,即T细胞起主要作用。它在受到结核菌的抗原刺激后可转化为致敏的淋巴细胞。当再次与结核杆菌相遇时,致敏的淋巴细胞可很快分裂、增殖,并释放出各种淋巴因子,如巨噬细胞趋化因子、集聚因子、移动抑制因子和激活因子等。这些因子可使巨噬细胞移向结核杆菌,并聚集于该处不再移动,这样就能把结核杆菌限制在局部不致扩散。同时还激活了巨噬细胞,使巨噬细胞体积增大,伪足形成活跃,溶酶体含量增加,细胞内pH下降等。这些改变有助于使吞入的细菌更易被水解、消化和杀灭。此外,激活后的T细胞还可释放其他淋巴因子,加强这一免疫反应,如结核杆菌的生长抑制因子能通过巨噬细胞特异性地抑制细胞内结核杆菌的繁殖而获得免疫。结核结节的形成就是上述各种反应的具体形态学表现。
结核病时发生的变态反应属于IV型(迟发性)变态反应。结核菌素试验就是这种反应的表现,本质上亦为细胞免疫反应。
结核病免疫反应和变态反应常同时发生并相伴出现,但两者关系如何及其对结核病的发生、发展有何影响等问题,长期来尚未解决。在结核病的诊断方面,基因诊断作为结核菌的非培养诊断技术是近年来结核病快速诊断的一项重大突破。它乃借分析结核菌的遗传物质核酸而特异,敏感、快速地检测和鉴定结核杆菌。目前用于结核菌检测和鉴定的基因诊断方法有基因探针技术、染色体指纹技术和PCR技术。
【结核病的基本病变】
结核杆菌在机体内引起的病变属于特殊性炎症,虽其病变具有一般炎症的渗出、坏死和增生三种基本变化,但有其特异性。由于机体的反应性(免疫反应和变态反应)、菌量及毒力和组织特性的不同,可出现以下不同的病变类型:
1.渗出为主的病变出现在结核性炎症的早期或机体免疫力低下,菌量多、毒力强,或变态反应较强时,表现为浆液性或浆液纤维素性炎。早期病灶内有中性粒细胞浸润,但很快被巨噬细胞取代。在渗出液和巨噬细胞内易查见结核杆菌。此型变化好发于肺、浆膜、滑膜和脑膜等处,说明与组织结构特性亦有一定的关系。渗出性变化可完全吸收不留痕迹,或转变为以增生为主或以坏死为主的病变。
2.增生为主的变化当菌量较少,毒力较低或人体免疫反应较强时,则发生以增生为主的变化,形成具有一定诊断特征的结核结节(结核性肉芽肿)。单个结核结节肉眼不易看见,三、四个结节融合成较大结节时才能见到。其境界分明,约粟粒大小,呈灰白半透明状,有干酪样坏死时则略呈黄色,可微隆起于器官表面。
结核结节(tubercle)是在细胞免疫基础上形成的,由类上皮细胞(epithelioid cell)、Langhans巨细胞加上外围局部集聚的淋巴细胞和少量反应性增生的纤维母细胞构成。当有较强的变态反应发生时,结核结节中便出现干酪样坏死。巨噬细胞体积增大逐渐转变为类上皮细胞,呈梭形或多角形,胞浆丰富,染淡伊红色,境界不清。核呈圆或卵圆形,染色质甚少,甚至可呈空泡状,核内可有1~2个核仁。多数类上皮细胞互相融合乃形成Langhans巨细胞,为一种多核巨细胞,体积很大,直径可达300μm,胞浆丰富,核与类上皮细胞核的形态大致相同,核数由十几个到几十个不等,有超过百个者。核排列在胞浆的周围呈花环状、马蹄形或密集在胞体的一端(图18-1)。
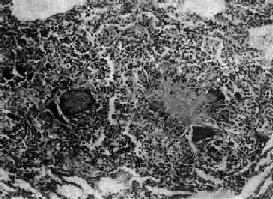
图18-1 结核性肉芽肿
结节中央为干酪样坏死,周围绕有类上皮细胞、Langhans巨细胞以及淋巴细胞等
3.坏死为主的变化在结核杆菌数量多、毒力强,机体抵抗力低或变态反应强烈的情况下,上述渗出性和增生性病变均可继发干酪样坏死。病变一开始便呈现干酪样坏死者十分少见。由于坏死组织含脂质较多(脂质来自破坏的结核杆菌和脂肪变性的单核细胞)而呈淡黄色,均匀细腻,质地较实,状似奶酪,故称干酪样坏死。镜下见为红染无结构的颗粒状物。干酪样坏死的形态特点,特别是肉眼所见对结核病的病理诊断具有一定的意义。干酪样坏死物中大都含有一定量的结核杆菌。
干酪样坏死灶内含有多量抑制酶活性的物质,故坏死物可不发生自溶、排出,也不易被吸收。但有时也能发生软化和液化,形成半流体物质。随着液化,结核杆菌大量繁殖,更进一步促进液化的发生。液化固有利于干酪样坏死物的排出,但更重要的是可成为结核菌在体内蔓延扩散的来源,是结核病恶化进展的原因。
以上渗出、增生和坏死三种变化往往同时存在而以某一种改变为主,而且可互相转化。例如渗出性病变可因适当治疗或机体免疫力增强而转化为增生性病变;反之,在机体免疫力下降或处于较强的变态反应状态时,原来的增生性病变则可转变为渗出性、坏死性病变,或原来的渗出性病变转化为坏死性病变。因此,在同一器官或不同器官中的结核病变是复杂多变的。
【结核病基本病变的转化规律】
结核病变的发展和结局取决于机体抵抗力和结核菌致病力之间的矛盾关系。当人体抵抗力增强时,细菌逐渐被控制而消灭,结核病变转向愈复;反之,则转向恶化。
1.转向愈复主要表现为病变的吸收消散、纤维化、纤维包裹和钙化。
(1)吸收消散:为渗出性病变的主要愈复方式。渗出物逐渐通过淋巴道吸收,病灶缩小或完全吸收消散。较小的干酪样坏死灶和增生性病变如治疗得当也可被吸收。
(2)纤维化、纤维包裹及钙化:增生性结核结节转向愈复时,其中的类上皮细胞逐渐萎缩,结节周围的增生纤维母细胞长入结核结节形成纤维组织,使结节纤维化。未被完全吸收的渗出性病变也可通过机化而发生纤维化。小的干酪样坏死灶(1~2mm)可完全纤维化;较大者难以完全纤维化而由坏死灶周围的纤维组织增生,将干酪样坏死物质加以包裹,以后干酪样坏死逐渐干燥浓缩,并有钙质沉着而发生钙化。
病灶发生纤维化后,一般已无结核杆菌存活,可谓完全痊愈。在被包裹、钙化的干酪样坏死灶中仍有少量细菌存活,病变只处于相对静止状态(临床痊愈),当机体抵抗力下降时病变可复燃进展。
2.转向恶化主要表现为病灶扩大和溶解播散。
(1)病灶扩大:病变恶化进展时,在病灶周围出现渗出性病变(病灶周围炎),其范围不断扩大,并继而发生干酪样坏死。坏死区又随渗出性病变的扩延而增大。
(2)溶解播散:干酪样坏死物发生溶解液化后,可经体内的自然管道(如支气管、输尿管等)排出,致局部形成空洞。空洞内液化的干酪样坏死物中含有大量结核杆菌,可通过自然管道播散到其他部位,引起新的病灶。如肺结核性空洞通过支气管播散可在同侧或对侧肺内形成多数新的以渗出、坏死为主的结核病灶。此外,结核杆菌还可通过淋巴道蔓延到淋巴结,经血道播散至全身,在各器官内形成多数结核病灶。
一、肺结核病
结核杆菌的感染途径主要是呼吸道,故结核病中最常见的是肺结核病。肺结核病可因初次感染和再次感染结核菌时机体反应性的不同,而致肺部病变的发生发展各有不同的特点,从而可分为原发性和继发性肺结核病两大类。
(一)原发性肺结核病
第一次感染结核杆菌所引起的肺结核病称原发性肺结核病,多发生于儿童,故又称儿童型肺结核病。但也偶见于未感染过结核杆菌的青少年或成人。
1.病变特点结核杆菌被吸入肺后,最先引起的病变称为原发灶。原发灶通常只有一个,偶尔也有二个甚至二个以上者。常位于通气较好的上叶下部或下叶上部靠近肺膜处。以右肺多见。病变开始时是渗出性变化,继而发生干酪样坏死,坏死灶周围有结核性肉芽组织形成。肉眼上,原发灶常呈圆形,直径多在1cm左右,色灰黄。结核杆菌很快侵入淋巴管,循淋巴流到所属肺门淋巴结,引起结核性淋巴管炎和淋巴结炎。表现为淋巴结肿大和干酪样坏死。肺的原发灶、淋巴管炎和肺门淋巴结结核三者合称为原发综合征(primary complex)(图18-2),是原发性肺结核病的病变特点。
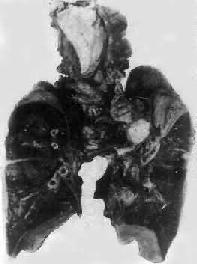
图18-2 肺结核病原发综合征
右侧肺膜下白色病灶为原发性,肺门部圆形白色病灶为干酪样变的肺门淋巴结
原发性肺结核病的症状轻微而短暂,常无明显的体征,很多患儿均在不知不觉中度过,仅表现结核菌素试验阳性。少数病变较重者,可出现倦怠、食欲减退、潮热和盗汗等中毒症状,但很少有咳嗽、咯血等呼吸道症状。
2.发展和结局 绝大多数(98%)原发性肺结核病患者由于机体免疫力逐渐增强而自然痊愈。小的病灶可完全吸收或纤维化,较大的干酪样坏死灶则发生纤维包裹和钙化(图18-3)。

图18-3 肺内钙化的肺结核原发灶
有时肺内原发病灶虽已愈合,而肺门淋巴结内的病变继续发展,结核菌通过淋巴道蔓延至附近淋巴结,使肺门附近更多的淋巴结受累,形成支气管淋巴结结核。经适当治疗后这些病灶仍可包裹、钙化而痊愈。
少数患儿在此时因营养不良或患其他传染病(如流感、麻疹、百日咳、白喉等),使机体抵抗力下降,病变因而恶化,肺内及肺门淋巴结病变继续扩大,并通过以下的途径播散(图18-4)。
图18-4 肺原发性结核病播散途径示意图
(1)淋巴道播散:肺门淋巴结病变恶化进展时结核杆菌可经淋巴管到达气管分叉处、气管旁、纵隔及锁骨上、下淋巴结引起病变(图18-5)。如果淋巴管因结核病变而被阻塞,结核菌则可逆流到达腹膜后及肠系膜淋巴结。预淋巴结亦可受累而肿大,此时喉头或扁桃体多有结核病灶存在。病变淋巴结肿大,出现干酪样坏死,并可互相粘连形成肿块。
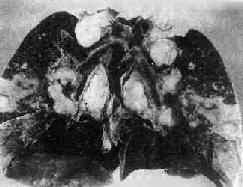
图18-5 肺结核原发综合征和肺门淋巴结结核
原发灶扩大,肺门淋巴结高度肿大,干酪化
(2)血道播散:结核杆菌侵入血流后经血道播散。若进入血流的菌量较少而机体的免疫力很强,则往往不致引起明显病变。如有大量细菌侵入血流,机体免疫力较弱时,则可引起血源性结核病。
(3)支气管播散:肺原发灶的干酪样坏死范围扩大,侵及相连的支气管,液化之坏死物质可通过支气管排出,形成空洞。同时,含有大量结核杆菌的液化坏死物还可沿支气管播散,引起邻近或远隔的肿组织发生多数小叶性干酪样肺炎灶。肺门淋巴结干酪样坏死亦可侵破支气管而造成支气管播散(图18-6)。但原发性肺结核病形成空洞和发生支气管播散者较少见。
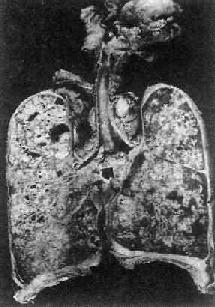
图18-6 肺结核支气管播散
图中箭头示干酪化的气管分叉淋巴结溃穿支气管,引起两侧小叶性干酪样肺炎及空洞形成
肺结核原发综合征恶化进展发生血道播散时,引起的血源性结核病有以下三种类型:
1)全身粟粒性结核病:当肺原发灶中的干酪样坏死灶扩大,破坏了肺静脉分支,大量结核杆菌由肺静脉经左心至大循环,可播散到全身各器官如肺、脑、脑膜、肝、脾、肾等处,形成粟粒性结核,称为急性全身性粟粒性结核病。肉眼见各器官内密布大小一致、分布均匀、灰白带黄、圆形的粟粒大小之结核结节。镜下,可为含菌较少的增生性病变,也可为含菌很多的渗出、坏死性病变。临床上,病情危重,有高热、衰竭、食欲不振、盗汗、烦躁不安等明显中毒症状,肝脾肿大,常有脑膜刺激征。若能及时治疗预后仍属良好,少数病例可因结核性脑膜炎死亡。
在机体抵抗力极差或用大量激素、免疫抑制药物或细胞毒性药物后,可发生严重的结核性败血症,为最剧烈的急性血源性全身性结核病,患者常迅速死亡。尸检时,在各器官内见无数小坏死灶,灶内含很多结核杆菌,灶周几乎无细胞反应可见,因而有无反应性结核病之称。此种病人可出现类似白血病的血像,称类白血病反应(leukemoid reaction)。
2)肺粟粒性结核病:又称血行播散型肺结核病。急性粟粒性肺结核病常是全身粟粒性结核病的一部分。偶尔,病变也可仅局限于两侧肺内。这是由于支气管周围肺门或纵隔淋巴结干酪样坏死破入附近的静脉(如无名静脉、颈内静脉、上腔静脉),含大量结核菌的液化物经右心和肺动脉播散至双肺所引起。肉眼观,双肺充血,重量增加,切面暗红,密布灰白或灰黄色粟粒大小的结节,微隆起于切面(图18-7),并显露于肺膜表面。
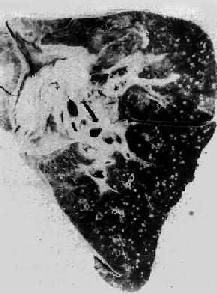
图18-7 肺粟粒性结核病
图中白色点状病灶为粟粒性结核灶
慢性粟粒性肺结核病多见于成年人,这时肺原发综合征业已钙化痊愈,结核杆菌由肺外(骨关节、泌尿生殖道及肾上腺等处)结核病灶较长期、间歇性地进入血流,播散于肺内,形成新旧不等的病变。间隔时间可为数月甚至数年。患者多因结核性脑膜炎死亡。
3)肺外器官结核病:或称肺外结核病,大多是原发性肺结核病经血道播散的后果。在原发综合征期间如有少量结核杆菌经原发灶内的毛细血管侵入血流,则能在肺外某些器官(骨关节,泌尿生殖器官、神经系统、浆膜、皮肤等)内形成个别的结核病灶。这些病灶可自愈或潜伏下来,经过较长时间后,当机体抵抗力下降时乃恶化进展为肺外器官结核病。
(二)继发性肺结核病
继发性肺结核病(secondary pulmonary tuberculosis)是指再次感染结核菌所引起的肺结核病,多见于成年人,故又称成人型肺结核病。肺内的病变常开始于肺尖,称再感染灶。关于再感染灶的形成机制有以下两种学说:①外源性再感染学说,认为继发性肺结核的发病是由外界重新感染所致,与原发性肺结核无任何联系;②内源性再感染学说,认为继发性肺结核病的再感染灶大多是由原发性肺结核病血源性播散时在肺尖部形成的病灶。在机体免疫力下降时,潜伏的病灶可发展为继发性肺结核病。此外,也可是肺内未愈合的原发灶内的结核菌经小支气管蔓延或由肺外器官结核病灶内的结核菌经血道播散至肺的结果。
继发性肺结核病患者对结核杆菌已有一定的免疫力或过敏性,所以继发性肺结核病与原发性肺结核病的病变有以下不同特点:①病变多从肺尖开始,这可能与人体直立位时该处动脉压低、血循环较差,随血流带去的巨噬细胞较少,加之通气不畅,以致局部组织抵抗力较低,细菌易在该处繁殖有关。②由于变态反应,病变发生迅速而且剧烈,易发生干酪样坏死,同时由于免疫反应较强,在坏死灶周围每有以增生为主的病变,形成结核结节。免疫反应不仅能使病变局限化,而且还可抑制细菌的繁殖,防止细菌沿淋巴道和血道播散,因此肺门淋巴结一般无明显病变,由血源播散而引起全身粟粒性结核病者亦极少见。病变在肺内蔓延主要通过受累的支气管播散。③病程较长,随着机体免疫反应和变态反应的消长,临床经过常呈波浪起伏状,时好时坏,病变有时以增生性变化为主,有时则以渗出、坏死变化为主,常为新旧病变交杂。
因此,继发性肺结核病的病变和临床表现都比较复杂。根据其病变特点和临床经过可分为以下几种主要类型:
1.局灶型肺结核 病变多位于肺尖下2~4cm处,右肺较多。病灶可为一个或数个,一般约0.5~1cm大小,多数以增生性病变为主,也可为渗出性病变,中央发生干酪样坏死。如病人免疫力较强,病灶常发生纤维化、钙化而痊愈。临床上病人常无明显自觉症状,多在体检时发现,属无活动性肺结核一类。如病人免疫力降低时,可发展成为浸润型肺结核。
2.浸润型肺结核是临床上最常见的一种类型,属于活动性肺结核。大多是局灶型肺结核发展的结果,少数也可一开始即为浸润型肺结核。病变中央常有较小的干酪样坏死区,周围有广阔的病灶周围炎包绕。镜下,肺泡内充满浆液、单核细胞、淋巴细胞和少数中性粒细胞,病灶中央常发生干酪样坏死。病人常有低热、盗汗、食欲不振、全身无力等中毒症状和咳嗽、咯血等。痰中常可查出结核杆菌。如能早期适当治疗,一般多在半年左右可完全吸收或部分吸收,部分变为增生性病变,最后,可通过纤维化、包裹和钙化而痊愈。
如病人免疫力差或未及时得到适当治疗,病变可继续发展,干酪样坏死灶扩大(浸润进展期)。坏死物质液化经支气管排出后形成急性空洞,洞壁粗糙不整,内壁坏死层中有大量结核杆菌,坏死层外可有薄层结核性肉芽组织包绕。从空洞中不断向外排出含菌的液化坏死物质,可经支气管播散,引起干酪样肺炎(溶解播散)。如靠近肺膜的空洞穿破肺膜,可造成自发性气胸;如果大量液化坏死物质进入胸腔,可发生结核性脓气胸。急性空洞一般较易愈合,如能给以及时和强有力的抗结核治疗,这种空洞可通过洞壁肉芽组织增生而逐渐缩小,最终形成瘢痕而治愈,或通过空洞塌陷,形成索状瘢痕愈合。若急性空洞经久不愈,则可发展为慢性纤维空洞型肺结核。
3.慢性纤维空洞型肺结核为成人慢性肺结核的常见类型,多在浸润型肺结核形成急性空洞的基础上发展而来。病变特点是在肺内有一个或多个厚壁空洞形成。同时在同侧肺组织,有时也可在对侧肺组织,特别是肺下叶可见由支气管播散引起的很多新旧不一、大小不等、病变类型不同的病灶,部位愈下病变愈新鲜。空洞多位于肺上叶,大小不一,呈不规则形,洞壁厚,有时可达1cm以上。洞内常见残存的梁柱状组织,多为有血栓形成并已机化闭塞的血管。空洞附近肺组织有显著纤维组织增生和肺膜增厚(图18-8)。镜下,洞壁分三层:内层为干酪样坏死物质,其中有大量结核杆菌;中层为结核性肉芽组织;外层为增生的纤维组织。由于病情迁延,病变广泛,新旧不等,肺组织遭到严重破坏,可导致肺组织的广泛纤维化,最终演变为硬化型肺结核,使肺体积缩小、变形、变硬、肺膜广泛增厚并与胸壁粘连,可严重影响肺功能。
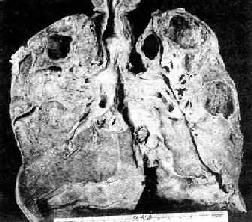
图18-8 慢性纤维空洞型肺结核病(冠状切面)
右上叶,左上叶上部和左下叶上部都有厚壁空洞形成;肺门淋巴结无结核病变
临床上,病程常历时多年,时好时坏。症状的有无与病变的好转或恶化相关。由于空洞与支气管相通,成为结核病的传染源,故此型有开放性肺结核之称。如空洞壁的干酪样坏死侵蚀较大血管,可引起大咯血,严重者可危及生命,病人多因吸入大量血液而窒息死亡。如空洞穿破胸膜可引起气胸或脓气胸。经常排出含菌痰液可引起喉结核。咽下含菌痰液可引起肠结核。肺广泛纤维化还可导致肺动脉高压,引起肺源性心脏病。
较小的结核空洞经过适当治疗可发生瘢痕愈合。较大的空洞经治疗后,洞壁坏死物质脱落净化,洞壁结核性肉芽组织逐渐转变为纤维瘢痕组织,与空洞邻接的支气管上皮增生并向空洞内伸延,覆盖于空洞内面。此时空洞虽仍存在,但已属愈合。空洞的这种愈合方式称为开发性愈合。
4.干酪样肺炎此种肺炎发生在机体免疫力极低,对结核菌的变态反应过高之病人,可由浸润型肺结核恶化进展而来,或由急、慢性空洞内的细菌经支气管播散所致。按病变范围大小的不同而分为小叶性和大叶性干酪样肺炎。后者可累及一个肺叶或几个肺叶。肉眼观,肺叶肿大变实,切面呈黄色干酪样,坏死物质液化排出后可见有急性空洞形成(图18-9)。镜下,肺泡腔内有大量浆液纤维素性渗出物,内含主为巨噬细胞的炎性细胞,且见广泛的干酪样坏死。抗酸染色可查见大量结核菌。此型结核病情危重,目前已很少见。
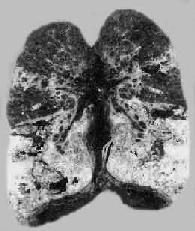
图18-9 干酪样肺炎
左肺下叶广泛性干酪化,肿大变实,数处形成边缘参差不齐的急性空洞。上叶亦被波及
5.结核球 又称结核瘤(tuberculoma),是孤立的有纤维包裹、境界分明的球形干酪样坏死灶,直径约2~5cm(图18-10)。多为一个,有时多个,常位于肺上叶。由于抗结核药物的广泛应用,结核球有明显增多的趋势。结核球可由浸润型肺结核转向痊愈时,干酪样坏死灶发生纤维包裹形成;亦可因结核空洞的引流支气管阻塞后,空洞由干酪样坏死物质填满而成;或由多个结核病灶融合而成。结核球为相对静止的病变,可保持多年而无进展,或发生部分机化和钙化而转向愈合,临床上多无症状。但亦可恶化进展,表现为干酪样坏死灶扩大、液化、溃破包膜、形成空洞和经支气管播散。因结核球干酪样坏死灶较大,周围又有纤维包裹,药物不易发挥作用,所以临床上多采取手术切除。

图18-10 肺结核球
为纤维包裹境界分明略呈同心圆层状的干酪样坏死灶
6.结核性胸膜炎在原发性和继发性肺结核病的各个时期均可发生,按病变性质可分为渗出性和增生性两种。
(1)渗出性结核性胸膜炎:较常见,大多发生于原发性肺结核病的过程中,且大多发生于原发综合征同侧胸膜。由肺原发灶或肺门淋巴结病灶中的结核菌播散至胸膜所引起,或为弥散在胸膜之结核菌体蛋白引起的过敏反应。患者多为较大的儿童或青年。病变主要表现为浆液纤维素性炎。浆液渗出量多时则引起胸腔积液,也可为血性胸水。当积液量不多,附有纤维素之胸膜壁层和脏层在呼吸时发生摩擦,可听到摩擦音。胸腔积液明显时,叩诊呈浊音,听诊时语颤和呼吸音减弱,并有肺受压及纵隔移位等体征。经积极治疗,一般可在1~2月后完全吸收而痊愈。如渗出物中纤维素较多,则可因发生机化而使胸膜增厚和粘连。
(2)增生性结核性胸膜炎:是由肺膜下结核病灶直接蔓延至胸膜所致。常发生于肺尖,多为局限性。病变以增生性变化为主,很少有胸腔积液。一般可通过纤维化而痊愈,并常使局部胸膜增厚、粘连。
二、肺外器官结核病
肺外器官结核病除淋巴结结核是由淋巴道播散所致、消化道结核可由咽下含菌的食物或痰液直接感染引起、皮肤结核可通过损伤的皮肤感染外,其他各器官的结核病多为原发性肺结核病血源播散所形成的潜伏病灶进一步发展的结果。肺外病变多数只限于一个器官内,常见有肠、腹膜、肾、生殖系、脑膜、骨关节等脏器,多呈慢性经过。
(一)肠结核病
肠结核病可分为原发性和继发性两型。原发性肠结核病很少见,常发生于小儿,一般是因饮用带有结核菌的牛奶而感染,并形成与原发性肺结核相同的原发综合征(肠的原发性结核性溃疡、结核性淋巴管炎及肠系膜淋巴结炎)。绝大多数肠结核继发于活动性空洞型肺结核病,因反复咽下含结核菌的痰液而引起。肠结核可发生于任何肠段,而以回盲部为其好发部位(约占85%),这是因为该段淋巴组织最为丰富,结核杆菌易于通过肠壁淋巴组织侵入肠壁;加之食物停留在回盲部的时间较长,接触结核杆菌的机会较多;这些因素均增加了结核病在该区发病的机会。其他部位如空肠下段,回肠与结肠肝曲、脾曲等处也可发生结核病变。依其病变特点的不同可分为两型:
1.溃疡型结核菌侵入肠壁淋巴组织引起结核结节形成,以后结节逐渐融合并发生干酪样坏死,破溃后形成溃疡。由于细菌随肠壁环形淋巴管播散,因此典型的肠结核性溃疡多呈带状,其长径与肠轴垂直(图18-11)。溃疡边缘参差不齐,底部附有干酪样坏死物质,其下为结核性肉芽组织。在肠的浆膜面每见纤维素和多数灰白色结核结节,连接成串,这是结核性淋巴管炎所致。溃疡愈合后常因疤痕形成和收缩引起肠腔狭窄。临床上可有腹痛、腹泻、营养障碍和结核中毒症状。
图18-11 结核性肠溃疡
溃疡呈横带状(半环形),其长径与肠轴垂直
2.增生型较少见。特点为肠壁内有大量结核性肉芽组织形成和纤维组织显著增生,肠壁高度肥厚变硬、肠腔狭窄。粘膜可有浅在溃疡或息肉形成(图18-12)。临床上表现为慢性不完全低位肠梗阻。右下腹常可扪及包块,易误诊为肠癌。
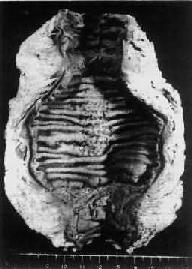
图18-12 肠增生型结核
肠壁内有大量结核性肉芽组织和纤维组织显著增生,使肠壁高度肥厚变硬,肠腔狭窄
(二)结核性腹膜炎
结核性腹膜炎多见于青少年,通常所见之慢性结核性腹膜炎大多继发于溃疡型肠结核、肠系膜淋巴结结核或结核性输卵管炎。由腹膜外结核灶经血道播散至腹膜者少见。可分为干、湿两型,但通常所见多为混合型。
1.湿型 腹膜充血,其上密布无数结核结节(图18-13),一般无腹膜粘连现象,或仅限于少数区域。腹腔内积存大量腹水,多呈草黄色,亦可为血性。病人常有腹胀、腹痛、腹泻及中毒症状。
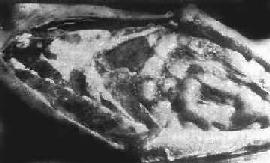
图18-13 结核性腹膜炎
腹膜上满布无数结核结节
2.干型其特点为腹膜上除见结核结节外尚有大量纤维素性渗出物,机化后引起腹腔器官,特别是肠管间、大网膜、肠系膜广泛紧密粘连。大网膜一般增厚、变硬和缩短。肠系膜也显著缩短。坏死严重者可在粘连的肠管之间或向腹外溃破形成瘘管。病人因肠粘连而出现慢性肠梗阻症状。腹上部可扪得一横行块状物,为收缩粘连的大网膜。并因腹膜增厚触诊时有柔韧感或橡皮样抗力。
(三)结核性脑膜炎
结核性脑膜炎以小儿多见,成人较少。主要由于结核杆菌经血道播散所致。在小儿往往是肺原发综合征血行播散的结果,故常为全身粟粒性结核病的一部分;在成人除肺结核病外,骨关节结核和泌尿生殖道结核病常是血源播散的根源。部分病例也可由于脑实质内的结核球液化溃破,大量结核菌进入蛛网膜下腔所致。
病理变化以脑底最明显。在脑桥、脚间池、视神经交叉及大脑外侧裂等处之蛛网膜下腔内,有多量灰黄色混浊胶冻样渗出物积聚。偶见比粟粒还小的灰白色结核结节(图18-14)。脑室脉络丛及室管膜有时也可有结核结节形成。镜下,蛛网膜下腔内炎性渗出物主要由浆液、纤维素、巨噬细胞、淋巴细胞组成,常有干酪样坏死,偶见典型结核结节形成。病变严重者可累及脑皮质而引起脑膜脑炎。病程较长则可发生闭塞性血管内膜炎,从而可引起多发性脑软化。未经适当治疗致病程迁延的病例,由于蛛网膜下腔渗出物的机化而发生蛛网膜粘连,可使第四脑室上中孔和外侧孔堵塞,引起脑积水。
图18-14 结核性脑膜炎
脑基底部脑膜增厚,有散在的结核结节
(四)泌尿生殖系统结核病
1.肾结核病 最常见于20~40岁。男性多于女性。多为单侧性、双侧性肾结核的发病率约为10%。病变开始于肾皮、髓质交界处或乳头体内。初为局灶性结核病变,病变继续扩大发展则发生干酪样坏死,破坏肾乳头而溃破入肾盂,成为结核性空洞。随着病变在肾内扩大蔓延,形成多数结核空洞,最后可使肾仅剩一空壳(图18-15)。由于液化的干酪样坏死物可随尿液下行,常使输尿管及膀胱感染。输尿管粘膜可发生溃疡和结核性肉芽组织形成,使管壁增厚、管腔狭窄,甚至阻塞,结果常引起肾盂积水或积脓。膀胱三角区往往最先受累,形成溃疡,以后可侵及整个膀胱,可引起膀胱壁纤维化,使膀胱容积缩小(膀胱挛缩)。膀胱的溃疡和纤维组织增生如影响到对侧的输尿管口,可使管口狭窄或失去正常的括约肌功能(关闭不全),造成对侧健肾引流不畅,最终可引起肾盂积水。结核菌也可逆行感染对侧肾。
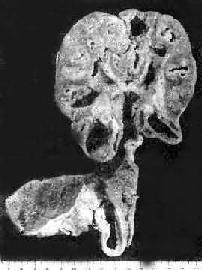
图18-15 肾结核及输尿管结核
肾实质内多数干酪样坏死灶及空洞形成;输尿管壁增厚,粘膜有多数结核结节及浅表溃疡形成
临床上,可因肾实质破坏而出现血尿。液化之干酪样坏死物在排出时可形成脓尿,尿中可查见结核菌。此外尚有尿频、尿急和尿痛。最初,这些症状是由于脓尿刺激膀胱所致,后来则是由于膀胱出现继发性结核之故。
2.生殖系统结核病男性生殖系统的结核病和泌尿系统结核病有密切关系。结核菌经过尿道可感染精囊和前列腺,以后可蔓延到输精管、附睾,睾丸偶尔也可受累。经血源感染引起之生殖系统结核病较少见。大多数病人双侧同时或先后发病。病变器官有结核结节形成和干酪样坏死。男性生殖系统结核的症状主要由附睾结核引起,前列腺和精囊结核多无明显症状。病侧附睾逐渐肿大,轻微疼痛或无痛,可与阴囊壁粘连,溃破后可形成长期不愈的窦道。
女性生殖系统结核多由血道或淋巴道播散而来,也可来源于邻近器官结核病的直接蔓延,以输卵管结核最多见,为女性不孕的原因之一。其次为子宫内膜,卵巢;子宫颈又次之。
(五)骨与关节结核病
骨、关节结核病多由血源播散所致,常见于儿童和青少年,因骨发育旺盛时期骨内血管丰富,感染机会较多。骨结核多侵犯脊椎骨,指骨及长骨骨骺(股骨下端和胫骨上端)等处。在关节结核中以侵犯髋、膝、踝、肘等关节多见。外伤可为发病诱因。
1.骨结核病变常由松质骨内的小结核病灶开始,以后病变的发展可分两型:比较多见者为干酪样坏死型,主要表现为明显的干酪样坏死,破坏骨质而开成死骨。病变常累及骨周围的软组织,引起干酪样坏死和结核性肉芽组织形成。坏死物质液化后可在骨旁形成结核性脓肿,局部并无红、热、痛,故又有冷脓肿之称。穿破皮肤后可形成经久不愈的窦道。另一种为增生型,比较少见。主要形成结核性肉芽组织,病灶内之骨小梁渐被侵蚀、吸收和消失,但无明显的干酪样坏死和死骨形成。以后病灶可为结缔组织包裹而静止。
脊椎结核是骨结核中最常见者。多侵犯第10胸椎至第2腰椎,少数见于颈椎。病变起于椎体,常发生干酪样坏死,病变发展可破坏椎间盘和邻近椎体。由于病变椎体不能负重,发生塌陷而呈楔形,造成脊柱后凸畸形(驼背)(图18-16)。若病变穿破骨皮质,可侵犯周围软组织,干酪样坏死物液化后可在局部形成结核性脓肿,或沿筋膜间隙向下流注,在远隔部位形成“脓肿”。由于脊椎后凸和椎旁结核性肉芽组织或“脓肿”压迫脊髓,可引起截瘫。
图18-16 脊椎结核
椎体和椎间盘干酪样坏死,造成椎体塌陷及后凸畸形
2.关节结核多继发于骨结核,病变通常开始于骨骺或干骺端,发生干酪样坏死。当病变发展侵入关节软骨和滑膜时则成为关节结核。关节滑膜内有结核性肉芽组织形成,关节腔内有浆液、纤维素性渗出物。游离的纤维素凝块长期互相撞击可形成白色圆形或卵圆形小体,称为关节鼠。关节附近的软组织呈水肿和慢性炎症,故关节部明显肿胀。病变累及软组织和皮肤时,可形成窦道。关节结核痊愈时,关节腔常被大量纤维组织充填,造成关节强直,失去运动功能。
(六)淋巴结结核病
最常受累的是颈部、支气管和肠系膜淋巴结,尤以颈淋巴结结核(俗称瘰疬)最为多见。病菌可来自肺门淋巴结结核的播散,亦可来自口腔、咽喉部结核感染灶。淋巴结常成群受累,有结核结节形成和干酪样坏死。淋巴结逐渐肿大,最初各个淋巴结尚能分离,当炎症累及淋巴结周围组织时,则淋巴结彼此粘连,形成较大的包块。颈淋巴结结核干酪样坏死物液化后可穿破皮肤,在颈部形成多数经久不愈的窦道。
第二节 麻风
麻风(leprosy)是由麻风杆菌引起的一种慢性传染病,主要病变在皮肤和周围神经。临床表现为麻木性皮肤损害、神经粗大,严重者甚至肢端残废。本病在世界上流行甚广,我国则流行于广东、广西、四川、云南以及青海等省、自治区。建国后由于积极防治,本病已得到有效的控制,发病率显着下降。
【病因及传染途径】
本病由麻风分支杆菌感染引起,从染色特性而论,麻风菌和结核杆菌一样,是一种抗酸菌。麻风菌的形态和结核菌相似,但较粗短,人工培养迄今尚未成功。本病的传染途径尚未十分明了,其传染源为瘤型和界限类患者,患者的鼻、口分泌液、汗液、泪液、乳汁、精液及阴道分泌物均含麻风菌,通常认为主要传染方式是藉上述分泌液或患者皮肤病变的微小伤口(甚至无破损的病变皮肤)排出的菌感染他人,一般系通过长期的直接接触,但间接接触(通过衣服、用具)可能也起一定作用。麻风杆菌侵入体内后,先潜伏于周围神经的鞘膜细胞或组织内的巨噬细胞内,受染后是否发病以及发展为何种病理类型,取决于机体的免疫力。对麻风杆菌的免疫反应以细胞免疫为主,患者虽亦有特异性抗体的产生,但抗体对抑制和杀灭麻风杆菌似不起重要作用。在细胞免疫力强的状态下,麻风杆菌将被巨噬细胞消灭而不发病,反之,麻风杆菌得以繁殖,引起病变。本病的潜伏期长达2~4年,但也有在感染数月后发病者。
麻风杆菌不能体外培养,使该菌表型鉴定困难。最近,用麻风杆菌特异核酸探针以点印迹杂交可分析活检组织中的麻风杆菌。PCR技术已被迅速应用于麻风病的诊断。
【病理变化】
由于患者对麻风杆菌感染的细胞免疫力不同,病变组织乃有不同的组织反应。据此而将麻风病变分为下述两型和两类:
1.结核样型麻风(tuberculoid leprosy) 本型最常见,约占麻风患者的70%,因其病变与结核性肉芽肿相似,故称为结核样麻风。本型特点是患者有较强的细胞免疫力,因此病变局限化,病灶内含菌极少甚至难以发现。病变发展缓慢,传染性低。主要侵犯皮肤及神经,绝少侵入内脏。
(1)皮肤:病变多发生于面、四肢、肩、背和臀部皮肤,呈境界清晰、形状不规则的斑疹或中央略下陷、边缘略高起的丘疹。镜下,病灶为类似结核病的肉芽肿,散在于真皮浅层,有时病灶和表皮接触。肉芽肿成分主要为类上皮细胞,偶有Langhans巨细胞,周围有淋巴细胞浸润(图18-17)。病灶中央极少有干酪样坏死,抗酸染色一般不见抗酸菌。因病灶多围绕真皮小神经和皮肤附件,故引起局部感觉减退和闭汗。病变消退时,局部仅残留少许淋巴细胞或纤维化,最后,炎性细胞可完全消失。
图18-17 结核样型麻风
真皮内有主由类上皮细胞构成的结节状病灶,其中可见Langhans细胞,颇似结核结节,但中央无干酪样坏死
(2)周围神经:最常侵犯耳大神经、尺神经、桡神经、腓神经及胫神经,多同时伴有皮肤病变,纯神经麻风而无皮肤病损者较少见。神经变粗,镜下有结核样病灶及淋巴细胞浸润。和皮肤病变不同的是神经的结核样病灶往往有干酪样坏死,坏死可液化形成所谓“神经脓肿”。病变愈复时类上皮细胞消失,病灶纤维化,神经的质地因而变硬。神经的病变除引起浅感觉障碍外,还伴有运动及营养障碍。严重时出现鹰爪手(尺神经病变使掌蚓状肌麻痹,使指关节过度弯曲、掌指关节过度伸直所致)、垂腕、垂足、肌肉萎缩、足底溃疡以至指趾萎缩或吸收、消失。在有效的防治措施下,上述肢体改变已不复见到。
2.瘤型麻风(lepromatous leprosy) 本型约占麻风患者的20%,因皮肤病变常隆起于皮肤表面,故称瘤型。本型的特点是患者对麻风杆菌的细胞免疫缺陷,病灶内有大量的麻风杆菌,传染性强,除侵犯皮肤和神经外,还常侵及鼻粘膜、淋巴结、肝、脾以及睾丸。病变发展较快。
(1)皮肤:初起的病变为红色斑疹,以后发展为高起于皮肤的结节状病灶,结节境界不清楚,可散在或聚集成团块,常溃破形成溃疡。多发生于面部、四肢及背部。面部结节呈对称性,耳垂、鼻、眉弓的皮肤结节使面容改观,形成狮容(facies leontina)。
镜下,病灶为由多量泡沫细胞(foamy cell)组成的肉芽肿,夹杂有少量淋巴细胞。泡沫细胞来源于巨噬细胞,在吞噬麻风杆菌后,麻风杆菌的脂质聚集于巨噬细胞浆内,乃使后者呈泡沫状。抗酸染色可见泡沫细胞内含多量麻风杆菌,甚至聚集成堆,形成所谓麻风球(globus leprosus)。病灶围绕小血管和附件,以后随病变发展而融合成片,但表皮与浸润灶之间有一层无细胞浸润的区域(图18-18),这是结核样型麻风所没有的。由于患者对麻风杆菌的细胞免疫缺陷,病灶内不出现类上皮细胞,淋巴细胞也很少。经治疗病变消退时,麻风杆菌数量减少,形态也由杆状变为颗粒状,泡沫细胞减少或融合成空泡,纤维组织增生。最后病灶消退仅留瘢痕。
图18-18 瘤型麻风
表皮萎缩变薄,真皮内有泡沫细胞的弥漫浸润,后者与表皮层间有一薄层无细胞浸润区相隔
(2)周围神经:受累神经也变粗,镜下,神经纤维间的神经束衣内有泡沫细胞和淋巴细胞浸润,抗酸染色可在泡沫细胞和Schwann细胞内查得多量麻风杆菌。晚期,神经纤维消失而被纤维瘢痕所代替。神经病变的临床表现和结核样型相似。
(3)粘膜:鼻、口腔,甚至喉和阴道粘膜均可受累,尤以鼻粘膜最常发生病变。
(4)脏器:肝、脾、淋巴结和睾丸等脏器常被瘤型麻风波及,可伴有肝、脾和淋巴结的肿大。镜下皆见泡沫细胞浸润。睾丸的曲细精管如有泡沫细胞浸润,可使精液含有麻风菌而通过性交传染他人。
3.界限类麻风(borderline leprosy) 本型患者免疫反应介于瘤型和结核样型之间,病灶中同时有瘤型和结核样型病变,由于不同患者的免疫反应强弱不同,有时病变更偏向结核型或更偏向瘤型。在瘤型病变内有泡沫细胞和麻风菌。
4.未定类麻风本类是麻风病的早期改变,病变非特异性,只在皮肤血管周围或小神经周围有灶性淋巴细胞浸润。抗酸染色不易找到麻风菌。多数病例日后转变为结核样型。少数转变为瘤型。
麻风的早期诊断和分型有赖于活体组织检查。及时的有效治疗可治愈。
第三节 伤寒
伤寒(typhoid fever)是由伤寒杆菌引起的一种急性传染病。病变主要特点是全身单核吞噬细胞系统的巨噬细胞反应性增生,尤以回肠淋巴组织的改变最为明显。临床上主要表现为持续性高热、神智淡漠、相对缓脉、脾肿大、皮肤玫瑰疹及血中白细胞减少等。
【病因及传染途径】
伤寒杆菌属沙门氏菌属,革兰染色阴性,呈短粗杆状,体周满布鞭毛,运动活泼,在含有胆汁的培养基中生长较好,因胆汁中的类脂及色氨酸可作为伤寒杆菌的营养成分。伤寒杆菌的菌体(O)抗原、鞭毛(H)抗原和表面(Vi)抗原能使人体产生相应的抗体。由于O及H抗原的抗原性较强,故可用于血清凝集试验(肥达反应,Widal reaction),以测定血清中的O及H抗体的效价来辅助临床诊断。菌体裂解时可释放强烈的内毒素,是伤寒杆菌致病的主要因素。目前利用沙门菌的invA基因和鞭毛素基因用PCR方法扩增进行分子杂交,可以检出3~300个活菌细胞,达到敏感和特异的效果。
伤寒患者和带菌者是本病的传染源。病菌随粪便和尿排出体外,通过污染饮水和食物,经口感染。苍蝇在本病的传播上起媒介作用。
【发病机制】
伤寒杆菌随污染的饮水或食物进入消化道后,穿过小肠粘膜上皮细胞侵入肠壁的淋巴组织,特别是回肠下段的集合淋巴小结和孤立淋巴小结,并沿淋巴管至肠系膜淋巴结。在这些淋巴组织内,伤寒杆菌一方面被巨噬细胞吞噬,并在其中生长繁殖;另一方面经胸导管进入血液,引起菌血症。血液中的病菌很快被全身单核吞噬细胞系统如肝、脾,骨髓和淋巴结中的巨噬细胞吞噬,并进一步在其中大量繁殖。在这一段时间内,虽然有单核吞噬细胞系统的增生反应,但临床上无明显症状,称为潜伏期,一般10天左右。
此后,在全身单核吞噬细胞系统内繁殖的病菌及其释放的内毒素再次大量进入血液,并随之散布至全身各脏器和皮肤等处,引起败血症和毒血症,呈现全身中毒性症状和病理改变。病变主要发生于回肠末段,其肠壁的淋巴组织出现明显的增生肿胀,此时相当于疾病的第一周,血培养常为阳性。随着病程的发展,在发病后的第2~3周,伤寒杆菌在胆囊内繁殖到一定数量,大量病菌随胆汁再度进入小肠,又可穿过肠粘膜再次侵入肠道淋巴组织,使原已致敏的肠壁淋巴组织发生强烈的过敏反应,导致坏死、脱落和溃疡形成。伤寒杆菌随同脱落的坏死组织和粪便排出体外,故此段时间粪便培养易获阳性结果。与此同时,人体的免疫力逐渐增加,血中的抗体不断上升,肥达反应在病程第二周以后多数出现阳性。但近年来有研究证明血中抗体滴度的高低与患者对伤寨杆菌的抵抗力无关,而系细胞免疫在对抗病菌上起主要作用。即在致敏T细胞所产生的某些淋巴因子的作用下,增强巨噬细胞的吞噬、灭菌功能。在病程的第四周,随着免疫力的增强,血液和器官内的细菌逐渐消失,中毒症状减轻、消失,病变随之愈合而告痊愈。
【病变及临床病理联系】
伤寒是主要累及全身单核吞噬细胞系统的感染性疾病,病变突出表现在肠道淋巴组织,肠系膜淋巴结、肝、脾和骨髓等处。此外,由于败血症的存在,在病菌及其释放的内毒素作用下,全身许多器官也可受累。
伤寒杆菌引起的炎症属急性增生性炎症,主要是巨噬细胞的增生。其吞噬能力十分活跃,胞浆中常吞噬有伤寒杆菌、受损的淋巴细胞、红细胞及坏死细胞碎屑,在病理诊断上具有一定的意义,故常称这种细胞为伤寒细胞。伤寒细胞常聚集成团,形成小结节,称为伤寒肉芽肿或伤寒小结(图18-19)。革兰染色可见伤寒细胞胞浆内含有被吞噬的伤寒杆菌。伤寒杆菌引起的炎性反应的特点是病灶内无中性粒细胞渗出。
图18-19 伤寒肉芽肿模式图
图示伤寒小结内大量增生的伤寒细胞,胞浆内含有红细胞及组织碎屑等
1.肠道病变以回肠下段的集合和孤立淋巴小结的病变最为常见和明显。按病变自然发展过程可分为以下四期,每期约1周。
(1)髓样肿胀期:在起病的第一周,回肠下段淋巴组织明显肿胀,凸出于粘膜表面,色灰红,质软。其中以集合淋巴小结肿胀最为突出,表面形似脑回样隆起(图18-20)。肠粘膜有充血、水肿、粘液分泌增多等变化。

图18-20 伤寒髓样肿胀期
图中可见肿胀的集合淋巴小结及孤立淋巴小结
(2)坏死期:从发病的第二周开始进入坏死期,肿胀的淋巴组织在中心部发生多数灶性坏死,并逐步融合扩大,累及粘膜表层。坏死组织失去正常光泽,色灰白或被胆汁染成黄绿色。
(3)溃疡期:一般发生于发病后第三周。由于坏死组织逐渐崩解脱落、形成溃疡。溃疡边缘稍隆起,底部高低不平。(图18-21)溃疡一般深及粘膜下层,坏死严重者可深达肌层及浆膜层,甚至穿孔,如侵及小动脉,可引起严重出血。

图18-21 伤寒溃疡期
图中可见三个椭圆形溃疡,边缘稍隆起,溃疡的长轴与肠的长轴平行
(4)愈合期:相当于发病后的第四周。溃疡面坏死组织已完全脱落干净,并长出肉芽组织将溃疡填平,然后由溃疡边缘的上皮再生覆盖而告愈合。
由于上述肠道病变,临床上每有食欲减退、腹部不适、腹胀、便秘或腹泻及右下腹轻压痛。粪便细菌培养在病程第二周起阳性率逐渐增高,在第3~5周阳性率最高可达85%。
由于临床早期应用有效抗生素如氯霉素,以上四期的病变极不典型。
2.其他单核吞噬细胞系统的病变
(1)肠系膜淋巴结:回肠下段附近的肠系膜淋巴结常显著肿大,充满大量吞噬活跃的巨噬细胞,也可有伤寒肉芽肿和灶性坏死形成。
(2)脾:中度肿大,约为正常的2~3倍,包膜紧张。切面呈混浊的暗红色,质甚软,有时如果酱样,并可用刀背刮下,脾小体不清楚。镜下见巨噬细胞弥漫性增生,并可有伤寒肉芽肿和灶性坏死形成。临床上在发病后第六天左右可触及肿大的脾,质软并具压痛。
(3)肝:肝肿大,质软。肝细胞呈高度浊肿,并有散在、边界较清的伤寒肉芽肿形成,肝窦扩张充血,汇管区可见单核细胞和淋巴细胞浸润。肝细胞也可因细菌毒素的直接作用或伤寒小结压迫引起的缺血而发生灶性坏死。
(4)骨髓:也有巨噬细胞增生、伤寒肉芽肿和灶状坏死形成。由于骨髓中的巨噬细胞摄取病菌较多,存在时间较长,故骨髓培养阳性率可高达90%,较血培养为高。
3.其他脏器的病变
(1)胆囊:虽然伤寒杆菌易在胆汁中大量繁殖,但大多数患者胆囊无明显病变或仅有轻度炎症。值得注意的是,病人临床痊愈后,细菌仍可在胆汁中生存,并通过胆汁由肠道排出,在一定时期内仍是带菌者,有的患者甚至可成为慢性带菌者或终身带菌者。
(2)心肌:心肌纤维有较重的混浊肿胀。重症患者可出现中毒性心肌炎。毒素对心肌的影响或毒素导致的迷走神经兴奋性增高,或许是临床上出现特征性重脉或相对缓脉的原因。
(3)肾:肾曲管上皮可发生混浊肿胀,以往认为临床上出现的蛋白尿与此有关。但近年来通过肾活检免疫荧光检查,发现肾小球毛细血管壁可有免疫球蛋白(IgG,IgM)及补体(C3)沉着,并查见Vi抗原,因此考虑为免疫复合物性肾炎,但这种肾的病变可迅速消退。尿培养早期多为阴性,病程第3~4周的阳性率约为25%。
(4)皮肤:部分患者在病程第7~13天,皮肤出现淡红色小斑丘疹,称玫瑰疹,以胸、腹及背部为多,一般在2~4天内消失。在皮疹中可查见伤寒杆菌。
(5)肌肉:膈肌、腹直肌和股内收肌常发生凝固性坏死(亦称蜡样变性)。临床出现肌痛和皮肤知觉过敏。
【并发症】
1.肠出血和肠穿孔均多发生于溃疡期。肠出血严重者可引起出血性休克。肠穿孔是伤寒最严重的并发症,穿孔多为一个,有时也可多个,且发生在肠胀气和腹泻的情况下,穿孔后常引起弥漫性腹膜炎。这两种并发症的发生率约为1%~5%。
2.支气管肺炎以小儿患者并发为多,常因抵抗力下降,继发肺炎球菌或其他呼吸道细菌感染所致,极少数病例也可由伤寒杆菌直接引起。
3.其他伤寒杆菌可借血道感染其他器官,如骨髓、脑膜、肾(肾实质及肾盂),关节,但皆少见。胆囊的感染常见且重要,因胆囊炎若成慢性,将长期排出伤寒菌作为本病的传染源。
【结局】
在无并发症的情况下,一般经过4~5周就可痊愈,病后可获得较强的免疫力。败血症、肠出血和肠穿孔是本病重要的死亡原因。自从使用抗生素治疗伤寒以来,病程显著缩短,临床症状也大为减轻,典型的伤寒肠道各期的病变及全身病变已属少见,但复发率却有一定的增加。
第四节 细菌性痢疾
细菌性痢疾(bacillary dysentery)是痢疾杆菌引起的一种常见肠道传染病。全年均可发生,但以夏秋季为多见。儿童发病率一般较高,其次是20~39岁青壮年,老年患者较少。
【病因及传染途径】
痢疾杆菌是革兰染色阴性的短杆菌。按抗原结构和生化反应可分为四群。福氏菌(Shigella flexneri)、宋内氏菌(Sh.sonnei)、鲍氏菌(Sh.boydii)和志贺氏菌(Sh.dysenteriae)。所有痢疾杆菌均能形成内毒素,志贺氏菌除内毒素外,还可产生外毒素。现用Ipa质粒抗原和志贺毒素(ShT)基因作PCR测定,为快速诊断开辟了新的徐径。
菌痢患者和带菌者是本病的传染源。痢疾杆菌从粪便中排出后,可直接或间接(通过苍蝇等)污染食物、饮水、食具、日常生活用具和手等,再经口传染给健康人。食物和饮水的污染有时可引起菌的暴发流行。
【发病机制】
痢疾杆菌经口进入消化道后,在抵抗力较强的健康人可被胃酸大部分杀灭,即使有少量未被杀灭的病菌进入肠道,亦可通过正常肠道菌群的拮抗作用将其排斥。此外,在有些过去曾受感染或隐性感染的患者,其肠粘膜表面有对抗痢疾杆菌和特异性抗体(多属分泌性IgA),能排斥痢疾杆菌,使之不能吸附于肠粘膜表面,从而防止菌痢的发生。而当人体全身及局部抵抗力降低时,如一些慢性病、过度疲劳、暴饮暴食及消化道疾患等,即使感染小量病菌也容易发病。
痢疾杆菌侵入肠粘膜上皮细胞后,先在上皮细胞内繁殖,然后通过基底膜侵入粘膜固有层,并在该处进一步繁殖,在其产生的毒素作用下,迅速引起炎性反应,其强度与固有层中的细菌数量成正比,肠上皮细胞坏死,形成溃疡。菌体内毒素吸收入血,引起全身毒血症。
中毒型菌痢大多发生于儿童,其发病机制尚未查明,可能因患者为特异体质,故对细菌毒素呈强烈过敏反应。
【病理变化及临床病理联系】
菌痢的病理变化主要发生于大肠,尤以乙状结肠和直肠为重。病变严重者,整个结肠甚至回肠下段也可受累。根据肠道炎症特征、全身变化和临床经过的不同,菌痢可分为以下三种:
1.急性细菌性痢疾病变初期呈急性卡他性炎,表现为粘液分泌亢进,粘膜充血、水肿、点状出血、中性粒细胞及巨噬细胞浸润,粘膜上皮坏死脱落后形成表浅糜烂。粘膜下层也可见炎性反应,但程度较轻。病变进一步发展乃成为本病特征性的假膜性炎,表现为粘膜表层坏死,同时在渗出物中出现大量纤维素,后者与坏死组织、中性粒细胞、红细胞和细菌一起形成假膜(图18-22)。假膜首先出现于粘膜皱襞的顶部,呈糠皮状,随着病变扩展可融合成片。假膜一般呈灰白色,如出血严重或被胆色素浸染时,则可分别呈暗红色或灰绿色(图18-23)。大约在发病后一周左右,在中性粒细胞破坏后释出的蛋白溶解酶作用下,纤维素和坏死组织发生溶解液化,而使假膜成片脱落,形成大小不等、形状不一的溃疡。溃疡多数浅表,甚少穿破粘膜肌层,但亦偶有深达肌层引起穿孔导致腹膜炎者。当病变趋向愈复时,肠粘膜的渗出物和坏死物逐渐被吸收、排出,组织的缺损经再生而修复。浅小的溃疡愈合后无明显瘢痕形成,深而较大的溃疡愈合后可形成浅表瘢痕,很少引起肠腔狭窄。
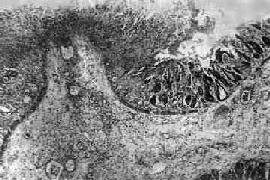
图18-22 细菌性痢疾
结肠粘膜表层坏死并有白细胞和纤维素性渗出物

图18-23 细菌性痢疾
结肠粘膜表面有假膜形成
肠系膜淋巴结偶可呈急性炎症而肿大。脾轻度肿大,白髓中细胞增生。肝、肾、心、脑等器官的实质细胞可发生变性甚至灶性坏死。
临床上,本病由于毒血症,可出现发热、头痛、乏力、食欲减退等全身症状和白细胞增多;由于病变肠管蠕动亢进并有痉挛,引起阵发性腹痛、腹泻等症状。由于炎症刺激直肠壁内的神经末梢及肛门括约肌,导致里急后重和排便次数频繁。随着肠炎症的变化,最初为稀便混有粘液,待肠内容物排尽后即转为粘液脓血便,偶尔排出片状假膜。排便每天10~20次或更多,但因量少,故脱水不明显。严重病例,大便次数频繁乃至失禁,并常伴有呕吐,可引起明显脱水、酸中毒和电解质紊乱、血压下降,甚至发生休克。
急性菌痢的自然病程为1~2周,在适当的治疗下大多痊愈,少数可转为慢性菌痢。并发症一般少见,包括肠出血、肠穿孔和关节炎等。后者每为多发性浆液性关节炎,多见于膝、踝、肘、腕等大关节。其发生可能为对感染的一种变态反应。
2.中毒型细菌性痢疾本型的特征为起病急骤,肠病变和症状常不明显,但有严重的全身中毒症状。发病后数小时或10数小时即可出现中毒性休克或呼吸衰竭。本型多见于2~7岁儿童,常由毒力较低的福氏或宋氏痢疾杆菌引起,由毒力强的志贺氏菌引起者反而少见。肠病变一般较轻,主要为粘液分泌增加、粘膜充血、水肿和少量中性粒细胞浸润等卡他性肠炎的改变。有时肠壁集合淋巴小结和孤立淋巴小结滤泡增生肿大,而呈滤泡性肠炎的变化。临床上常无明显的腹痛、腹泻及脓血便。
3.慢性细菌性痢疾菌痢病程超过2个月以上者称为慢性菌痢。多由急性菌痢转变而来,其中从福氏菌感染转为慢性者为多。慢性菌痢的病程可长达数月或数年,在此期间随着患者全身及局部抵抗力的波动,肠道病变此起彼伏,新旧混杂,原有病损尚未完全愈合,而新的病损又可发生,此时,可能是由于肠壁神经装置受损,使再生修复过程障碍而有慢性溃疡形成。此种慢性溃疡边缘不规则,边缘粘膜常过度增生而形成息肉。溃疡多深达肌层,底部高低不平,有肉芽组织和瘢痕形成。由于肠壁反复受损的结果,纤维组织大量增生,使肠壁增厚,严重者可造成肠腔狭窄。
临床上可出现不同程度的肠道症状,如腹痛、腹胀、腹泻或便秘与腹泻交替出现,经常带有粘液或少量脓血。在急性发作肠的炎症加剧时,可出现急性菌痢的症状。大便培养痢疾杆菌有时阳性,有时阴性。有少数慢性菌痢患者可无明显症状和体征,但大便培养持续阳性,成为慢性带菌者,常为传播菌痢的传染源。
第五节 钩端螺旋体病
钩端螺旋体病(leptospirosis)是由钩端螺旋体引起的一种急性传染病。此病几遍及世界各大洲,尤以热带和亚热带为着。我国已有28个省、市、自治区发现本病,并以盛产水稻的中南、西南、华东等地区流行较重。发病季节主要集中在夏秋(6~10月)水稻收割期间,常以8~9月为高峰,青壮年农民发病率较高。在气温较高的地区,终年可见其散发病例。
【病因】
钩端螺旋体属密螺旋体,一般长6~10μm,宽0.1~0.2μm,有12~18个螺旋,其一端或两端常弯曲成钩
■[此处缺少一些内容]■
方法可以从脑脊液,尿,血查出钩端螺旋体DNA,达到特异、敏感、快速诊断、并可区分类型。
【传染途径】
鼠类和猪是本病的主要传染源。农业劳动者,接触污染的田水,钩端螺旋体常经皮肤(特别是破损的皮肤)进入人体,引起本病的流行。在洪水泛滥或大雨后,也可有本病的流行,主要是猪的含菌排泄物污染水源所致。此外,污染的水或食物亦可经消化道粘膜引起感染;在患本病的孕妇,钩端螺旋体还可经过胎盘使胎儿受染。
【疾病发展过程及其发病机制】
由于钩端螺旋体菌型、毒力大小和侵入人体数量的不同,以及人体免疫力和反应性的强弱,病理改变和临床表现的轻重及类型亦有很大差异。依疾病的发展过程及其发病机制其病程可概括为三个阶段。
1.早期(败血症期)此期约相当于起病后的1~3天,为钩端螺旋体侵入人体后,经淋巴道和小血管进入血循环和组织内大量繁殖并产生毒素所致。病理形态改变比较轻微,而功能改变较为显著。主要是全身感染中毒性微血管功能改变及肺、肝、肾、心、中枢神经系统等急性功能障碍。临床表现为早期中毒症候群,如畏寒、发热、乏力、头痛、躯干痛、结膜充血、腓肠肌压痛,表浅淋巴结肿大、皮疹和鼻衄等。
2.中期(败血症伴器官损伤期)此期多发生于起病后第3~10天。在败血症继续发展的基础上,钩端螺旋体及其毒素进一步引起不同程度的器官损害,造成临床上不同的病型。如无明显器官损害,临床上表现为流感伤寒型;如有明显器官损害,则根据受损器官及其严重程度,分别表现为肺出血型、黄疸出血型(又称Weil病)、肾功能衰竭型和脑膜炎型等。各型间有时可有一定重叠。
3.后期(恢复期或后发症期)此期多发生在起病第7~10天以后。钩端螺旋体侵入人体后,最初出现非特异性免疫反应,表现为肝、脾、淋巴结等单核吞噬细胞系统细胞增生,巨噬细胞和中性粒细胞增多,并不同程度地吞噬钩端螺旋体,但不能彻底予以消灭。在发病后1周左右,血液中开始出现特异性抗体,首先出现IgM,以后出现IgG,其水平随病程逐渐增高。与此同时,血液及各组织中的钩体开始减少并消失,临床上遂进入恢复期。多数患者热退后各种症状逐渐消失而获痊愈。少数患者在热退后几天至6个月或更长时间可再出现发热、眼部及神经系统后发症。可能为迟发性变态反应所致。
【病变及临床病理联系】
本病的病理变化属急性全身性中毒性损害,主要累及全身毛细血管,引起不同程度的循环障碍和出血,以及广泛的实质器官变性、坏死而导致严重功能障碍。炎性反应一般轻微。由于菌型和毒力的不同,所引起病变的轻重和主要累及器官的种类也有异。
1.肺主要表现为肺出血,在肺出血型患者最为显著,多为黄疸出血群所引起,为近年来无黄疸钩端螺旋体病患者常见的死亡原因,一般出现在病后3~5天,暴发者也可发生在病后1~2天。最初出血呈点状分布,以后点状出血不断增多和扩大,并互相融合,形成全肺弥漫性出血,使两肺各叶体积增大,重量增加,外表呈紫红色,有如肝。切面见肺叶几乎全部或大部呈出血性实变,色暗红,酷似血凝块。气管和支气管几乎全为血液所充满(图18-24)。镜下,肺泡壁毛细血管显著充血,管腔极度扩张,但未见明显血管破裂现象。支气管和肺泡内充满大量红细胞,肺水肿和炎性反应均不明显。
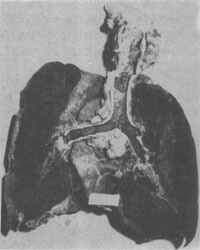
图18-24 弥漫性肺出血型钩端螺旋体病的肺切面几乎全部呈出血变实,酷似血凝块
2.肝肝的病变以黄疸出血型患者最为显著。肉眼观察肝肿大,质软,色黄。镜下见肝细胞混浊肿胀、脂肪变性和小叶中央灶性坏死。Disse腔水肿,肝细胞索离解。枯否细胞增生。汇管区胆小管可见胆汁淤滞和淋巴细胞、中性粒细胞及少量嗜酸性粒细胞浸润。钩体含量在全身各器官、组织中,以肝为最多。主要存在于Disse腔和毛细胆管内,肝细胞和枯否细胞内可见变性的钩端螺旋体。由于肝细胞的损害可引起胆汁排泄功能降低和凝血功能障碍,故临床上可见重度黄疸和广泛皮肤、粘膜出血。严重者可发生急性肝功能不全或肝肾综合征。
3.肾肾损害可以单独存在(肾型钩端螺旋体病),也可与流感伤寒型、肺出血型,特别是黄疸出血型同时存在。肉眼观,肾稍肿大,切面见皮质苍白,髓质淤血,偶见肾包膜出血。镜下主要见间质性肾炎和肾小管上皮细胞不同程度的变性坏死。肾小管腔扩大,其中含有蛋白、红细胞、粒细胞和细胞碎片,并可形成管型阻塞肾小管管腔。肾间质可见充血、水肿或出血,有散在的小灶性炎细胞浸润,主要为淋巴细胞、单核细胞、少量中性和嗜酸性粒细胞浸润。肾小球一般无明显改变。肾组织内易查见钩端螺旋体,可见于肾间质、肾小管上皮细胞及肾小管腔内。肾损害严重者可引起急性肾功能不全,造成肾损害的原因除毒素作用外,肾微循环障碍所致的缺氧亦起一定的作用。
4.其他器官组织病变
(1)心脏:心脏常扩大,质地较软,心外膜和心内膜可见出血点。镜下见心肌细胞普遍混浊肿胀,偶见灶性坏死。间质有水肿、出血和血管周围炎,以单核细胞浸润为主,夹杂有少数中性粒细胞和淋巴细胞。临床上可出现心动过速、心律紊乱及心肌炎的征象。
(2)横纹肌:以腓肠肌病变最为明显。镜下主要见肌纤维节段性变性、肿胀、横纹模糊或消失,并可出现肌浆空泡或溶解性坏死,肌浆及肌原纤维溶解消失,仅存肌纤维轮廓。间质有水肿、出血和少量炎细胞浸润。临床上出现腓肠肌压痛。
(3)神经系统:部分病例有脑膜及脑充血、出血、炎细胞浸润和神经细胞变性。这些病变在钩体病的脑膜脑炎型最为明显。临床上出现脑膜脑炎的症状和体征。
(4)脾、淋巴结:单核巨噬细胞增生和炎细胞浸润,可引起脾和淋巴结肿大。
(5)肾上腺:皮质内脂质减少或消失,皮质和髓质均可有出血及灶性或弥漫性炎细胞浸润。
【后发症】
1.眼部病变眼部病变多在热退后1周至1月左右发生。最常见者为虹膜睫状体炎、脉络膜炎或全葡萄膜炎。并可出现巩膜表层炎,球后神经炎或玻璃体混浊。
2.脑动脉炎患者多为儿童,亦可为青壮年。患者常在急性期症状消失后1~5月出现偏瘫和失语等症状。主要病变是脑底多发性动脉炎及其所引起的脑实质损害。病变主要累及脑底动脉各干支,以大脑中动脉及大脑前动脉病变最重,大脑后动脉次之。病变呈跳跃性节段性分布,使该动脉粗细不匀、僵直或呈灰白色绳索状。此外,在基底节及其邻近脑实质内可见毛细血管及小静脉明显扩张充血,有的聚集成簇。镜下见受累动脉早期可为全动脉炎,较晚期则主要为增生性动脉内膜炎,使内膜显著增厚,可引起血管腔狭窄甚至闭塞,偶尔可并发血栓形成(图18-25)。脑实质可因缺血而发生轻重不一和不同部位的水肿、出血和多发性软化灶。
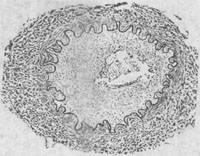
图18-25 钩端螺旋体病后发症——脑动脉炎模式图动脉内膜高度增生变厚,管腔变狭窄
第六节 性传播疾病
性病指通过性行为传染而又在社会上有较重要流行病学意义的疾病。这类疾病包括梅毒、淋病、性病性淋巴肉芽肿(lymphogranuloma venereum,又称腹股沟淋巴肉芽肿,lymphogranuloma inguinale)、腹股沟肉芽肿(granuloma inguinale,又称性病性肉芽肿,granuloma venereum)、软性下疳(chancroid)等。实际上通过性行为传染而又能在社会上造成流行性影响的疾病种类并不少,包括衣原体引起的非淋病性尿道炎、结膜炎、宫颈炎,支原体引起的非淋病性尿道炎,单纯疱疹病毒引起的外阴疱疹,外阴疣病毒引起的外阴尖锐湿疣,阴道滴虫引起的滴虫性尿道炎以及人类免疫缺陷病毒引起的艾滋病等,然而通常所谓的性病多只指梅毒、淋病、性病性淋巴肉芽肿、腹股沟肉芽肿和软性下疳等。本节仅叙述梅毒和淋病。
一、梅毒
梅毒(syphilis)是苍白密螺旋体(Treponema pallidum,亦称梅毒螺旋体)引起的一种慢性传染病,通常通过性交传染,其传播和社会因素有非常密切的关系。本病的特点是病程的长期性和潜匿性,病原体可侵犯任何器官,临床表现出各种不同的症状,也可陷匿多年而毫无临床表现。
【病因及传播方式】
梅毒螺旋体常在直接接触破损的皮肤或粘膜时才能进入人体。本病95%以上经性交传播,少数可因输血、接吻、医务人员不慎受染等直接接触传播,这二种传播方式所获得的梅毒称为后天性梅毒(获得性梅毒)。梅毒螺旋体可自患病母体的血液经胎盘传给胎儿,引起先天性梅毒(胎传梅毒)。新生儿神经梅毒,用PCR对患儿CSF检查是证实梅毒螺旋体感染的唯一方法。
【发病机制和基本病变】
患者对梅毒螺旋体感染后会产生细胞免疫和体液免疫。免疫力强弱决定受染后是痊愈、潜匿,抑或发展为晚期梅毒。在本病的较晚阶段(晚期第二期梅毒以后),患者对该病原菌的抗原发生细胞介导的迟发性变态反应,使病原体所在部位形成肉芽肿(树胶肿)。患者也产生体液免疫,在感染后第6周血清出现特异性抗体,有血清诊断学意义。患者可有肾病综合征的临床表现,其病理基础为膜性肾小球肾炎,肾小球毛细血管基底膜上皮下有免疫复合物沉积。对患者重要器官的破坏作用而言,细胞介导的迟发性变态反应所引起的树胶肿最为重要。
梅毒的基本病变有二:一为灶性闭塞性动脉内膜炎及血管周围炎,后者表现为围管性单核细胞、淋巴组织和浆细胞浸润。浆细胞的恒定出现是本病的特色之一。另一为类似结核的肉芽肿,该肉芽肿韧而有弹性,质地如树胶,故称树胶肿(gumma)。树胶肿灰白色,大小颇为悬殊,大者达数厘米,小者仅能见于镜下。其镜下结构颇似结核结节,中央为干酪样坏死,弹力纤维染色尚可见到组织内原有的血管壁轮廓,类上皮细胞和Langhans巨细胞则较少。树胶肿久后可被吸收,纤维化,最后形成使器官变形的瘢痕收缩,但绝少钙化,这又和结核结节截然有别。树胶肿可发生于任何器官,最常见于皮肤、粘膜、肝、骨和睾丸。血管炎病变能见于各期梅毒,而树胶肿则见于第三期梅毒。
【病程分期】
本病通过性交传染,病原体由皮肤或粘膜侵入,其病程发展经过下述三阶段:
第一期梅毒梅毒螺旋体侵入人体内后,约经10~90天,通常约3周左右的潜伏期,在侵入的局部发生充血,水疱,水疱不久溃破,形成质硬、基底洁净、边缘耸起的溃疡,称为下疳(chancre),因基质硬,乃称硬性下疳,以和Ducrey嗜血杆菌引起的另一种性病——软性下疳相区别。下疳通常发生于阴茎头、阴唇和子宫颈,约10%的病例发生于生殖器以外,如唇、舌、肛周。数目为一个。镜下改变为溃疡底部闭塞性动脉内膜炎和血管周围炎。特殊染色(Levaditi染色或免疫荧光染色)可检见螺旋体。约30%~50%的患者,其下疳太小可不被察觉,或从未发生明显的溃疡。下疳发生约1周后,局部淋巴结肿大,镜下呈非特异性急性或慢性炎。
及时治疗可阻止向第二期梅毒发展,但由于患者产生的免疫反应,即使不加治疗,下疳也可于2~6周后自行愈合,肿大的局部淋巴结消退。
第二期梅毒第一期梅毒如不治疗,即使下疳愈合,潜伏于体内的螺旋体仍能继续繁殖,在感染后第8~10周左右大量进入血循环,引起全身广泛性皮肤粘膜病变,即梅毒疹(syphilid)。通常表现为口腔粘膜红斑、丘疹、躯干、四肢、掌心和足心的斑疹、丘疹和口唇、外阴、肛周的扁平湿疣(condyloma lata)。后者表现为暗红色突起的平坦斑块。所有梅毒疹的组织学变化皆为淋巴细胞和浆细胞浸润构成的非特异性炎及闭塞性血管内膜炎和血管周围炎(图18-26),扁平湿疣则尚有表皮增生和角化不全。凡梅毒疹病灶内皆有苍白螺旋体。第二期梅毒有全身性淋巴结肿大,镜下为非特异性炎。少数患者可发生亚急性脑膜炎、虹膜炎或肝炎,以及免疫复合物反应所引起的膜性肾小球肾炎。梅毒疹也可不治“自愈”,但患者实际陷入隐性梅毒阶段,若不治疗,多年后30%的患者将发生第三期或晚期梅毒。第二期梅毒若予治疗,将阻止其向第三期梅毒发展。
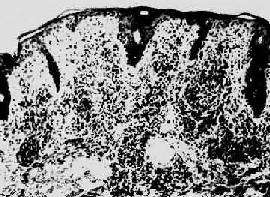
图18-26 梅毒疹
真皮呈致密的淋巴细胞及浆细胞浸润(采自Muir)
第三期梅毒第三期梅毒和第一、二期梅毒之不同在于其破坏性病变,即干酪样坏死和瘢痕形成。病变最常发生于心血管(80%~85%),次为中枢神经(5%~10%),此外肝、骨骼等器官也常发生,实际上几乎任何器官皆可受累。
1.心血管梅毒病变主要发生于动脉(梅毒性主动脉炎)。早在第一期梅毒时,螺旋体已经血道或淋巴道栖止于主动脉壁,潜伏达15~20年之久,才发生病理性损害,是以患者通常皆为40~55岁之中年人。梅毒性主动脉炎的特征为病变起始于主动脉升部,遍及主动脉弓及胸主动脉,每于横膈段截然而止。初始病变为主动脉外膜滋养血管的闭塞性内膜炎,由于管腔的逐渐闭塞,导致主动脉中层弹性纤维和平滑肌的缺血和退行性变,继而由瘢痕取代。镜下,主动脉外膜血管周围有围管性淋巴细胞和浆细胞浸润,小血管内膜增生,中层有灶性微小瘢痕和淋巴细胞、浆细胞浸润(图18-27)。微小的树胶肿偶见。弹性纤维染色可见主动脉中层弹力组织变薄,并有散在的小瘢痕取代了原有的弹力纤维(图18-28)。肉眼观,由于瘢痕收缩及内膜的纤维增生,使内膜表面呈弥漫分布的微细而深陷的树皮样皱纹。又因弹力纤维的广泛破坏,主动脉可呈梭形或囊状扩张,称为主动脉瘤(aortic aneurysm)。患者可因主动脉瘤破裂而猝死。主动脉梅毒的又一病变为主动脉瓣的瓣膜环部病变所致的后果:①环部弹力纤维破坏引起瓣膜环部扩张,②瓣膜呈纤维增生,③瓣叶联合处由于主动脉壁内膜纤维增生和瓣膜收缩而发生瓣叶间的分离。三项因素综合导致主动脉瓣关闭不全。梅毒性主动脉瓣病变的特点是瓣叶之间绝无粘连,故关闭不全而不伴有狭窄,这是和风湿性主动脉瓣病变不同之处。主动脉瓣关闭不全,使左心室异常肥大和扩大,以致心重可达千克左右,乃有“牛心”(car bovinum)之称。患者最终死于心力衰竭。此外,主动脉根部瘢痕又造成冠状动脉口狭窄,导致心绞痛发生。第三期梅毒的心血管病变主要涉及主动脉,仅偶有心肌发生树胶肿者。

图18-27 梅毒性主动脉炎
主动脉中层滋养血管周围有致密的圆形细胞浸润(采自Muir)

图18-28 梅毒性主动脉炎
中层弹力组织变薄、断裂,内膜增厚(右上角)弹力纤维染色(采自Muir)
2.中枢神经梅毒病变涉及中枢神经及脑脊髓膜,有脑膜血管梅毒、脊髓痨(tabes dorsalis)和麻痹性痴呆(paretic dementia)。脑膜血管梅毒病变主要发生于脑底,表现为脑膜血管周围和血管外膜的淋巴细胞和浆细胞浸润。肉眼观,脑膜混浊,部分病例可伴有脑膜小动脉内膜增厚,引起脑皮质多发性梗死。脑膜血管的病变持续发展可演变成麻痹性痴呆。这时脑膜增厚,脑膜小动脉闭塞更趋严重而普遍,导致脑皮质及中央灰质缺血、梗死,伴有胶质细胞增生。巨噬细胞和皮质血管壁有含铁的色素为本病的特征。病变处脑皮质(以额叶最为显著)萎缩,并呈颗粒状,脑室也可扩张,室管膜增厚,呈颗粒状。脊髓痨为脊髓后束(薄束和楔束)变性,肉眼可见后束萎缩,有时病变了波及后根。脊髓膜增厚,伴有淋巴细胞及浆细胞浸润。本病病变以脊髓末段受侵犯最早也最严重。
3.其他器官常见的病变为树胶肿,肝的树胶肿可使肝呈结节状肿大,树胶肿的干酪样坏死吸收和纤维化时,随着瘢痕收缩,肝变为分叶状,称分叶肝(hepar lobatum)。骨的树胶肿可导致骨折,睾丸树胶肿临床可误诊为肿瘤。
【先天性梅毒】
受梅毒螺旋体感染的妇女受孕时,胎儿可通过胎盘而被感染,称为先天性梅毒(congenital syphilis)。受梅毒感染2~5年间的孕妇,其胎儿的受染率最大。早在胎龄2~3月时,胎儿体内已有螺旋体存在,但先天性梅毒病变只见于胎龄4个月以上的胎儿及嗣后出生的婴幼儿。先天性受染的胎儿可死于胎内,引起晚期流产,可为死产,或出生不久即死亡。轻度感染可待发育到儿童期或青年期始发病。
胎内及新生儿先天性梅毒的突出病变为皮肤和粘膜的广泛大皰和大片的剥脱性皮炎,内脏的淋巴细胞和浆细胞浸润、动脉内膜炎、弥漫性纤维化和发育不全等。肺、肝呈弥漫性纤维化。胰、脾、心肌也有类似病变。骨软骨炎为恒见之病变,表现为长骨骨骺线的梅毒性肉芽肿形成,从而破坏软骨骨化过程。鼻骨和硬腭可因树胶肿的破坏形成马鞍鼻(saddle nose)和硬腭穿孔。凡上述病变,皆易在组织切片内查出苍白螺旋体。
婴儿先天性梅毒病变以皮肤粘膜病变和骨软骨炎为主,内脏有纤维化和梅毒性肉芽组织形成。内脏病变程度决定病婴存活期的久暂。长骨的骨膜炎伴有骨膜的新骨生成,胫骨前侧骨膜的增生使胫骨向前呈弧形弯曲,形成所谓马刀胫(saber shin)。马鞍鼻、眼、脉络膜炎和脑膜炎亦甚常见。
发生于2岁以上幼儿的梅毒为晚发性先天性梅毒,临床特征除间质性角膜炎、马刀胫和马鞍鼻外,由于牙发育障碍,中切牙小而尖,又由于牙釉质发育不全,中切牙切缘呈镰刀状缺陷,称为Hutchinson齿。内脏器官也可有类似于获得性梅毒第三期的病变,脑膜血管梅毒常侵犯神经和视神经。间质性结膜炎、Hutchinson齿和神经性耳聋构成晚期先天性梅毒的三联征。
二、淋病
淋病(gonorrhea)是淋球菌(N.gonorrhoeae)引起的急性化脓性炎。在男性,病变开始于前尿道,可逆行蔓延至后尿道,波及前列腺、精囊和附睾;在女性,病变可累及外阴和阴道的腺体、子宫颈粘膜以及输卵管。本病的危害性不仅在于它在所有性病中最为常见,而且根据统计,近年来其发病率在一些国家显着上升。这是因为本病有极强的传染性,患者并可为无症状的带菌者,在当今淋球菌对青霉素、四环素、链霉素已有抗药性的情况下,更易不受控制地感染他人。
本病通过性交传染,首先感染前尿道引起尿道炎,也可借污染的手指或毛巾感染结膜。淋球菌有毒性不大的内毒素,但内毒素的致病作用不明。患者可对该内毒素产生免疫反应,但也仅为一时性。淋球菌表面的多糖成分有抗吞噬作用,但若一旦被白细胞吞噬,淋球菌迅即死亡。淋球菌的基因探针诊断可以快速地检测和鉴定淋球菌。
【病理变化】
成人外阴和阴道的鳞状上皮能抗拒淋球菌的侵入,但淋球菌可侵入前尿道粘膜和尿道的附属腺体。在受染的第2~7天,尿道和尿道附属腺体的急性卡他性化脓性炎使脓性渗出物自尿道口流出,尿道口充血、水肿。如不治疗则病变上行延及后尿道及其附属腺体、前列腺、附睾和精囊腺,或Batholin腺、Skene腺(女性的尿道旁腺)、子宫颈以至输卵管,引起上述组织的化脓性炎,然而难以解释的是睾丸炎却极少见。在前列腺可形成脓肿。之后,尿道炎性瘢痕导致尿道狭窄,排尿困难。输卵管炎可导致输卵管繖粘连,脓性渗出物积于输卵管腔内成为输卵管积脓,从而局部感染慢性化。输卵管病变如延及卵巢,两者粘连,形成输卵管卵巢脓肿;病变更可扩展至盆腔,引起盆腔器官粘连。上述病变可导致所谓盆腔炎性疾病(pelvic inflammatory disease,一种含义并不十分明确的临床病理综合征,大部分病例为淋病所致)。患者可因而不育。
约1%~3%的患者可发生菌血症。临床上似脑膜炎球菌败血症的暴发性败血症也可发生,但极少见,表现为皮肤的点状出血和脓包,甚至发生急性心内膜炎和脑膜炎。
近年来基因诊断技术的应用,使淋病快速诊断有重大突破。目前用于淋球菌检测的基因诊断方法主要是基因探针技术。基因探针有质粒DNA探针,菌毛DNA探针,染色体基因探针和rRNA基因探针。取少许尿道或宫颈分泌物提取DNA,经PCR扩增,然后用其探针检测,具有敏感性高,特异性强的优点。
第七节 深部真菌病
真菌引起的疾病称为真菌病。真菌种类很多,与细菌相比,对人致病者相对较少,约150种。近年来由于广谱抗生素、肾上腺皮质激素和免疫抑制剂的大量应用,真菌感染有明显增长。真菌病根据病变部位不同可分成浅部真菌病和深部真菌病两大类。浅部真菌病主要侵犯含有角质的组织,如皮肤、毛发和指甲等处,引起各种癣病。深部真菌病侵犯皮肤深层和内脏,如肺、脑、消化道等器官,危害性较大。
真菌一般不产生外毒素,其致病作用可能与真菌在体内繁殖引起的机械性损伤以及所产生的酶类、酸性代谢产物有关。真菌及其代谢产物具有弱抗原性,在人体内可引起变态反应导致组织损伤。真菌的致病力一般较弱,与其致病力有关的因素是感染量和毒力,感染途径和机体免疫状态。只有当机体抵抗力降低时才能侵入组织,大量繁殖引起疾病。造成机体抵抗力降低,诱发深部真菌病的因素有以下几种:①慢性消耗性疾病,如恶性肿瘤、糖尿病和尿毒症等,可使机体免疫功能和抵抗力降低;②长期使用广谱抗生素,敏感的细菌被抑制,消除了正常菌群中与真菌相拮抗的细菌,使真菌得以大量繁殖。有些抗生素可促进某些真菌的生长,如金霉素可促进念珠菌生长;③肾上腺皮质激素可抑制炎症反应和纤维细胞增生,并可稳定细胞内溶酶体的膜,阻止其中酶的释放,影响中性粒细胞和巨噬细胞溶解杀灭微生物的作用。肾上腺皮质激素还能破坏淋巴细胞,使抗体形成减少;④大剂量X线照射、抗肿瘤药物和免疫抑制剂都能抑制骨髓,使中性粒细胞和巨噬细胞减少,机体的免疫功能和抵抗力降低。这些药物并可损伤正常组织和细胞,引起消化道粘膜坏死、出血,为真菌侵入创造条件;⑤治疗用的长时间静脉插管,留置导尿管和大手术,可引起局部损伤,成为真菌侵入的门户,在机体抵抗力低落时在体内繁殖致病。
诊断真菌病有四种基本方法:临床表现,真菌学、免疫学和病理学。最直接的方法是能证明真菌存在于组织中,或在渗出物中可分离培养。病理组织学的改变只有少数病变具有特征性。
一、念珠菌病
念珠菌病(candidiasis)是由念珠菌引起的一种常见的真菌病。念珠菌是一种酵母样菌,种类很多,在自然界中广泛存在。最常见的致病菌种为白色念珠菌(Candida albicans),常存在于正常人的口腔、皮肤、阴道和消化道内,因此念珠菌病多为内源性感染。念珠菌病可为急性或慢性,病变多样,可发生在身体各个部位。皮肤和粘膜的浅部念珠菌病较常见。温暖、潮湿环境有利于真菌生长,因此皮肤念珠菌病多发生在腋窝、腹股沟和指(趾)间、肛门周围等处。有时并可侵犯指甲引起甲床炎和甲沟炎。糖尿病人和孕妇发生念珠菌性外阴阴道炎者较多,可能与糖尿病人血糖和尿糖增高,以及孕妇阴道上皮细胞内糖原增多有关。
深部念珠菌病多为继发性,常继发于慢性消耗性疾病、体弱婴儿、严重的营养不良及恶性肿瘤等。在恶性肿瘤中,尤其是急性白血病并发念珠菌病者较多见。
【病理变化】
念珠菌引起的病变大致有三种:①在病变部经常有数量多少不等的单核细胞、淋巴细胞和中性粒细胞浸润,部分有小脓肿形成;②真菌侵犯的组织发生坏死,形成大小不等的坏死灶,其中炎性细胞较少;③肉芽肿性反应,这种病变比较少见,除一般炎性细胞浸润外,并有多核巨细胞和类上皮细胞形成结节状肉芽肿。以上病变可单独存在,也可同时发生。病变组织内并可见念珠菌的芽生孢子和假菌丝。念珠菌为圆形或椭圆形生芽的酵母样菌(图18-29),壁薄,直径约2~5μm。由芽管延长而形成的假菌丝细长而直(图18-30),有分隔,有时有少数分支。细长的假菌丝常侵入组织的深层,并可侵入血管(图18-31)。在组织切片内同时见到芽生孢子和假菌丝可诊断为念珠菌病。这些孢子和菌丝在HE染色切片中呈蓝色,如用革兰染色或镀银法则显示更为清晰。
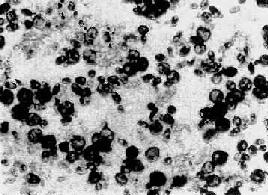
图18-29 胃念珠菌病
胃溃疡表面的念珠菌芽生孢子
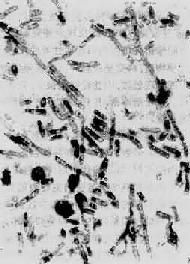
图18-30 胃念珠菌病
念珠菌的假菌丝(此图与图18-29为同一切片)

图18-31
胃溃疡底部可见细长的假菌丝侵入血管(此图与图18-29同一切片)
皮肤和粘膜念珠菌感染常在皮肤和粘膜表面形成不规则的白色片状假膜状物。口腔粘膜的念珠菌病称为鹅口疮。全身性念珠菌病多发生在消化道和呼吸道、心、肾、脑、肝、脾等处。消化道的念珠菌以食管下端和胃最多见。粘膜表面有灰白色假膜,并常有溃疡形成。粘膜内和溃疡底部有大量真菌孢子和菌丝,常向深部生长,侵入粘膜下层和肌层。支气管和肺念珠菌病常继发于肺结核病和支气管扩张症,临床症状与支气管炎和肺炎相似,并可形成小脓肿,晚期可引起纤维化和肉芽肿形成。临床上对支气管及肺念珠菌病的诊断必须审慎,单纯从痰中分离出念珠菌不能作为念珠菌病的诊断根据。因念珠菌常存在于正常人痰及唾液中,故需多次培养阳性,再结合其他表现才能确定诊断。
肾和泌尿道念珠菌感染可引起肾盂肾炎。念珠菌性心内膜炎可发生于风湿性心瓣膜病变和人工心瓣膜手术后病人。损伤的心瓣膜上有大的赘生物形成。念珠菌可侵犯脑膜及脑,引起脑膜炎,并在脑组织内形成肉芽肿和小脓肿。念珠菌侵入血流可引起念珠菌性败血症和播散性念珠菌病,全身各脏器皆可受累,常为病人的致死原因。
二、隐球菌病
隐球菌病(cryptococcosis)是新型隐球菌(cryptococcus neoformans)引起的一种亚急性或慢性真菌病。最常见的是中枢神经系统隐球菌病。也可发生于肺、皮肤、骨和其他器官。
新型隐球菌是一种腐物寄生性酵母菌。可以从土壤、鸽粪和水果中分离出来,也可从健康人的皮肤、粘膜和粪便中分离出来。环境中的病原体主要通过呼吸道,也可通过皮肤或消化道进入人体引起疾病,或使成为带菌者。隐球菌病少数可为原发性,多数为继发性。后者以何杰金病和白血病病人并发隐球菌病者最多见。
【病理变化】
新型隐球菌为圆形或卵圆形和酵母菌,一般为单芽,厚壁,有宽阔、折光性的胶质样荚膜。隐球菌大小相差很大,不包括荚膜一般直径多在4~7μm左右。有些直径可达20μm。周围荚膜由粘多糖组成,厚约3~5μm。在HE染色组织切片中,隐球菌呈淡红色,不易察见。用PAS反应或爱先蓝染色则清晰可见。
隐球菌在组织内引起慢性炎症反应。病变与病期早晚有关,早期由于病原体产生大量荚膜物质,病变呈胶胨样。这些荚膜物质能抑制粒细胞的趋向性和吞噬作用,所以病灶内炎症反应轻微,中性粒细胞很少,只有少数淋巴细胞和组织细胞浸润。新鲜活动的病灶中可见大量隐球菌,并有多数呈出芽状态,悬浮于胶胨样荚膜物质中,有些病原体被巨噬细胞吞噬。
晚期病变为肉芽肿性。病变区域纤维组织增生,其间有大量巨噬细胞、异物巨细胞和淋巴细胞。多数巨噬细胞和异物巨细胞胞浆内都可有隐球菌。肉芽肿性病灶逐渐扩大可压迫周围组织,以后病灶可由纤维组织包围或形成纤维疤痕,一般不发生钙化。
中枢神经系统隐球菌病主要表现为脑膜炎。脑膜增厚,以脑底部最明显,蛛网膜下腔内有大量稠厚的胶胨样物质和少数巨噬细胞。有时细胞渗出物较多,并有肉芽肿形成。脑膜可与脑组织粘连,影响脑脊液循环。感染可沿血管周围间隙侵入脑组织,有时可在脑组织内(主要在灰质和基底节)形成许多小囊腔。镜下可见囊腔为扩大的血管周围间隙(图18-32),腔内充满隐球菌及其所排泌的胶样物质。其中的血管细小,有时可有血管内膜炎和血栓形成。周围脑组织可因缺血而发生软化。胶质细胞可有轻度增生。后期,肉芽肿性病灶可发生在脑膜、脑实质和脊髓。隐球菌性脑膜炎起病缓慢,临床症状与结核性脑膜炎相似,很容易误诊。脑实质病变范围较大时,常与脑的占位性病变混淆,脑脊液检查寻找病原体可确定诊断。
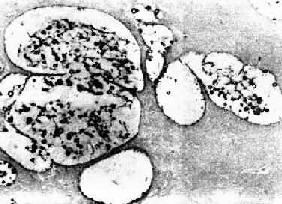
图18-32 脑隐球菌病
切片示脑组织内形成许多小囊腔,腔内充满隐球菌及其所分泌的凝胶样物质
许多隐球菌性脑膜炎病从都有肺部感染病史,并且有些隐球菌脑膜炎病人的肺部同时也有病变。因此,一般认为原发病灶在肺,病原体乃通过血道播散到脑、脑膜、皮肤、骨、内脏和身体其他部位;也有人认为鼻咽部隐球菌感染可通过淋巴道播散到脑。
肺隐球菌病可在肺组织内形成肉芽肿结节,大小不等,直径约1~8cm,可为单个或多个。多数在胸膜下形成单个小结节,有时可误诊为结核病或肺癌。镜下可见肉芽肿内有多数隐球菌和巨噬细胞。有时巨噬细胞排列在病灶周围甚似结核结节结构。有时病变为进行性,形成多数粟粒性肉芽肿结节和大的胶胨样病灶。
三、曲菌病
曲菌病(aspergillosis)由曲菌引起。曲菌是最常见的污染杂菌,种类很多,在人类曲菌病中,最常见的致病菌为烟曲菌(Aspergillus fumigatus)。曲菌生长迅速,在潮湿霉烂的谷物、稻草或腐烂的枯树叶中繁殖很快。因此谷仓、土壤、空气中常有大量曲菌孢子,可引起原发性肺曲菌病。外界环境中的曲菌孢子主要通过呼吸道进入人体。吸入的曲菌孢子不一定在人体内繁殖引起疾病。多数情况下曲菌是一种条件致病菌,只在机体抵抗力降低的基础上致病。
【病理变化】
曲菌可在身体许多部位引起病变,如皮肤、耳、鼻腔、眼眶、心、脑、肾、呼吸道、消化道等,而以肺部病变为最常见。曲菌在组织内可引起化脓,形成小脓肿。有时不化脓而发生组织坏死及出血,周围有多数中性粒细胞和单核细胞浸润。在小脓肿和坏死灶内有大量菌丝。曲菌并常侵入血管引起血栓形成,造成血管阻塞,可使组织缺血、坏死。慢性病灶有纤维组织增生,其中有大量淋巴细胞和单核细胞浸润,并杂有多数巨噬细胞和异物巨细胞。病灶内有大量菌丝。曲菌菌丝粗细均匀,直径约2~7μm,有隔,分支状,常成约45°的锐角分支(图18-33),HE染色呈蓝紫色,PAS染色更为清晰,也可用镀银法显示。在陈旧的纤维肉芽肿性病灶内,曲菌菌丝可变形,粗细不匀,部分可膨大呈球状或小囊状。
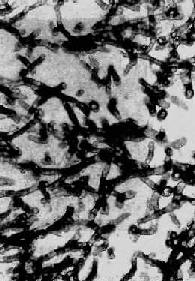
图18-33 曲菌病
曲菌菌丝粗细均匀,有隔,呈锐角分枝
支气管和肺的曲菌病表现为支气管炎或支气管肺炎。在支气管粘膜或肺组织引起小脓肿形成和肺梗死。曲菌常在肺组织原有的空洞性病变,如肺结核空洞、支气管囊性扩张或肺脓肿等的空腔内繁殖。大量菌丝形成团块,称为曲菌球。一般为发生在上叶或下叶尖段。曲菌球逐渐增大,并可在空腔内移动,可引起咯痰和反复咯血。
还有一种过敏性支气管肺曲菌病。临床表现类似支气管哮喘,痰粘稠,痰液内可分离出曲菌。血及痰内嗜酸性粒细胞增多。血清IgE增高,并有抗曲菌抗原的沉淀抗体。发病机制可能与曲菌抗原引起变态反应,使支气管痉挛、分泌亢进有关。主要病变为近端小支气管破坏,引起中心性或近端支气管扩张。晚期造成肺组织纤维化。过去认为这种过敏性支气管肺曲菌病很少见,但近年来发现在接触大量曲菌孢子的人员中并不少见,并逐渐增多,值得注意。此病早期治疗可以恢复,可防止发展到晚期引起严重的支气管扩张和肺纤维化。
鼻窦的曲菌病可直接蔓延到眼眶和脑及脑膜。脑及脑膜的曲菌病也可由血源播散而来。曲菌易侵犯血管,引起小动脉和静脉坏死性脉管炎。当病人抵抗力低下时,可引起血源播散到心肌、脑、肾、消化道、肝、脾等器官,形成小脓肿和肉芽肿性病变。曲菌性心内膜炎可在心房或心室内膜及瓣膜上形成大的赘生物。消化道曲菌病多发生在小肠、胃、食管和舌,其粘膜表面坏死,形成溃疡。
四、毛霉菌病
毛霉菌病(mucormycosis)由毛霉菌引起。与其他真菌病之多呈慢性经过不同,毛霉菌病多表现为急性炎症,发展很快,常引起广泛播散,并常侵袭血管引起血栓形成和梗死。尤其是脑毛霉菌病可在短期内造成死亡。
毛霉菌常在霉烂的水果、蔬菜、干草、肥料内大量繁殖。因此土壤、空气内常有大量毛霉菌。毛霉菌孢子在空气中飞扬可通过呼吸进入鼻窦和肺(图18-34)。有时并可随食物进入消化道。但毛霉菌极少在健康人中引起疾病,毛霉菌病几乎全为继发。
图18-34 肺毛霉菌病
【病理变化】
毛霉菌病的病变主要为急性化脓性炎症。毛霉菌侵袭性很强,很快向邻近组织扩散,并且常侵犯血管,引起血栓形成和血道播散。毛霉菌侵犯血管比曲菌严重,造成血管阻塞和梗死者也较多。因此,组织的严重坏死、化脓可能是毛霉菌直接作用和霉菌栓子阻塞血管所共同造成的后果。有时病变可较为慢性,可见多数巨噬细胞和异物巨细胞,其间并有多量中性和嗜酸性粒细胞浸润,间质纤维组织增生,毛细血管壁增厚。病变部分包括坏死区、血管壁、血管腔和血栓内都有大量毛霉菌菌丝。毛霉菌菌丝粗大,壁厚,直径约5~60μm,大多数直径在10~15μm之间。不分隔,有时可形成皱褶,易误认为分隔。分支较少而不规则,常呈钝角或直角分支(图18-35)。在组织内一般无孢子,菌丝在HE染色切片中容易被苏木素着色,因此明显可见。PAS反应效果反而不好。巨噬细胞和异物巨细胞胞浆内也可见吞噬的菌丝。
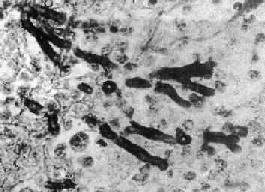
图18-35 毛霉菌病
毛霉菌菌丝粗大,无隔,分支少而不规则
头面部毛霉菌病最常见于糖尿病酸中毒时。病原体由鼻粘膜侵入,在鼻粘膜上形成黑色坏死灶,然后向周围扩散,可蔓延到硬腭引起粘膜溃疡,并可蔓延到同侧眼眶,累及眼球、眶内软组织、血管和神经。毛霉菌侵入眼动脉引起血栓形成可造成颊部、眼眶内容物和眼睑梗死以及局部皮肤坏疽。病原体可通过眼眶蔓延到颅腔,引起颈内动脉或海绵窦血栓、毛霉菌性脑膜炎和脑坏死。头面部毛霉病病情凶险,发展迅速,故早期诊断非常重要。当病人出现糖尿病酸中毒、单侧眼眶周围感染和脑膜脑炎三联症时要特别注意。此时可做鼻粘膜刮片培养或取粘膜做活检,检查毛霉菌。病原体侵入支气管粘膜,穿过支气管壁,侵犯肺门部组织、肺动脉和肺静脉,可引起肺动脉血栓形成和肺梗死。毛霉菌孢子随食物进入消化道,侵犯粘膜和血管可引起食管、胃和小肠坏疽、溃疡形成或穿孔。有时病变蔓延到肝、横膈或肠系膜。病原体由头颅部或肺部病灶侵入血流可引起全身播散,在多数器官形成急性炎症和栓塞性病变。
五、放线菌病
放线菌病(actinomycosis)主要是以色列氏放线菌(actinomyces israeli)引起的一种慢性化脓性炎症。根据现代的生物学分类,放线菌不属于真菌而属于一种厌氧细菌。由于它所引起的病变与霉菌病相似,以及按照过去的习惯,通常仍将放线菌病与真菌病一起叙述。放线菌有细胞壁,其化学成分与细菌相似,没有核膜。菌体呈细丝状,与真菌相似,菌丝的粗细与普通杆菌相似,一般直径不超过1μm。菌丝分支,可断裂为杆状。放线菌的种类很多。在自然界分布极广,空气、土壤、水源中都有放线菌存在。少数菌株对人类有致病性,其中最主要的为以色列氏放线菌。以色列氏放线菌是人口腔正常菌群中的腐物寄生菌,在拨牙、外伤或其它原因引起口腔粘膜损伤时,放线菌可由伤口侵入。也可通过吞咽或吸入带菌物质进入胃肠或肺。因此放线菌病主要发生于颈面部和胸腹器官。
【病理变化】
放线菌病和病变为慢性化脓性炎症。局部组织水肿,有大量中性粒细胞和单核细胞浸润,其间逐渐出现许多大小不等的坏死区,形成多数小脓肿,周围纤维组织增生。脓肿大小不等,常相互融合,并向邻近组织蔓延,形成许多窦道和瘘管。脓肿壁和窦道周围肉芽组织内有大量中性粒细胞、淋巴细胞和单核细胞浸润,有时并有少数多核巨细胞,部分可见大量吞噬脂类的巨噬细胞,因此肉眼观常带黄色。放线菌在脓肿壁、窦道壁和脓腔内繁殖,形成菌落。有时肉眼可见脓液内有细小的黄色颗粒,直径约1~2mm,称为“硫黄颗粒”。取硫黄颗粒直接压片或在组织切片中可见颗粒由分支的菌丝交织而成。在HE染色的组织切片中,颗粒中央部分染蓝紫色,周围部分菌丝排列成放线状,菌丝末端常有胶样物质组成的鞘包围而膨大呈棒状,染伊红色,所以称为放线菌(图18-36)。
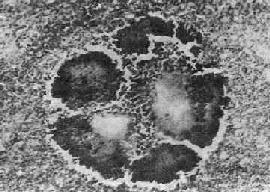
图18-36 放线菌病
病灶中的“硫黄颗粒”,周围部分菌丝排列成放线状。菌丝末端膨大呈棒状
有时组织切片中菌丝不明显,可作革兰染色,放线菌菌丝体为革兰阳性,胶样鞘为革兰阴性,据此可确诊为放线菌病。
放线菌病常同时合并其他细菌感染。病变常迁延不愈。一处病变纤维化,附近可出现新的病灶,再形成脓肿。日久后可引起大量组织破坏和瘢痕形成。颈面部放线菌病最多见,约占放线菌病总数的一半。病原菌常在口腔粘膜损伤(如拨芽等)时侵入。病变多发生在颌骨附近。早期,牙龈及邻近软组织肿胀、质硬、表面皮肤暗红色,以后液化形成脓肿。病变向周围组织扩展,形成多数脓肿,彼此沟通形成窦道,并常穿破皮肤形成瘘管。病变并可侵犯颌骨引起骨膜炎和骨髓炎,严重者可进一步扩展到颅骨、脑膜及脑。
腹内放线菌病多发生于阑尾和结肠,在粘膜下层形成小脓肿。病变常穿透肠壁引起局限性腹膜炎,并可侵入邻近肠袢、腹膜后组织和腹壁,形成排脓的窦道。有时并可通过淋巴管或血道、或直接蔓延到肝引起多发性肝脓肿,进一步可引起膈下脓肿,最后可破入胸腔引起胸腔内感染。感染也可沿腰肌蔓延到肾周围组织和腰椎,并可引起腰肌脓肿。
胸内放线菌病因吸入放线菌引起,或由腹部放线菌病蔓延而来,常形成肺脓肿,逐渐扩散可形成肺胸膜瘘或脓胸。进一步可侵犯胸壁及肋骨,引起胸壁瘘管。有时可蔓延到心包引起化脓性心包炎。
第十九章 寄生虫病
第一节 阿米巴病
阿米巴病(amoebiasis)是由溶组织内阿米巴(entamoeba histolytica)原虫感染引起,病变发生于结肠,在少数病例病原体可进一步移行到肝、肺和脑,偶尔蔓延到肛周皮肤、泌尿、生殖等器官。
本病遍及世界各地,但以热带及亚热带地区为多见。在我国多见于南方,但在夏季也常见于北方。由于我国卫生状况的不断改善,近年来本病的流行和急性病例已明显减少。
一、肠阿米巴病
肠阿米巴病是由溶组织内阿米巴寄生于结肠而引起的,因临床上常出现腹痛、腹泻和里急后重等痢疾症状,故常称为阿米巴痢疾。
【病因和发病机制】
肠道阿米巴病主要由于食入被包囊污染的食物和水而引起。包囊囊壁具有抗胃酸作用,能安全地通过胃而到达结肠的回盲部,在肠液的消化作用下脱囊而出,发育成为小滋养体。小滋养体直径约为3~12μm,以二分裂方式繁殖,吞噬肠内容物和细菌为营养,不能吞噬红细胞。在结肠功能正常时,横结肠以下的肠段内,因水分吸收,营养物减少和粪便增加,小滋养体停止活动,遂进入包囊前期,继而产生囊壁形成包囊,最后随粪便排出,故小滋养体也称为肠腔内型或共栖成囊型滋养体。大多数感染者为无症状的包囊携带者,是本病的重要传染源。溶组织阿米巴有共栖特点和潜在致病力,是否致病取决于虫体和宿主间的相互作用。虫株的毒力和侵袭力强,宿主局部肠功能紊乱、细菌感染、粘膜损伤或全身因营养不良、感染和免疫功能低下时,小滋养体便侵袭组织,发生致病作用。小滋养体粘附于结肠上皮,凭借其伪足的机械运动及其酶的溶解性破坏作用侵入肠壁,遂转变为大滋养体并大量增殖,造成局部肠粘膜溶解坏死和溃疡形成。大滋养体直径多为20~30μm,能活跃地吞噬红细胞和组织碎片,为致病型也称组织型滋养体。大滋养体可随坏死组织进入肠腔,排出体外后死亡,或在肠腔中转变为小滋养体,再形成包囊。
溶组织内阿米巴的致病机制目前尚不完全清楚,其毒力和侵袭力主要表现在对宿主组织的溶解破坏作用。实验证明,滋养体对宿主细胞的破坏具有接触溶解(contact lysis)的特点。滋养体表面有具溶酶体活性的膜结合酶(membrane bound enzyme),最近有人纯化一种膜结合的成孔蛋白(pore-forming protein),通过膜电位变化打开离子通路,使细胞因离子流失而死亡。从溶组织内阿米巴的纯培养中可分离出一种细胞毒素——肠毒素(enterotoxin),这种不耐热的蛋白质,可能在肠阿米巴病的粘膜损伤和腹泻中起重要作用。此外,超微结构研究认为,滋养体表面的丝状伪足可能具有吞噬宿主细胞或胞饮宿主物质,附着并钻入组织,释放细胞毒素及接触溶解宿主细胞等一系列作用。另外,需要指出的是,溶组织内阿米巴滋养体与肠道某些细菌的共生作用在协同致病上起重要作用,细菌不仅作为阿米巴的营养来源,亦为它们提供适宜的生长、繁殖环境和促进阿米巴的代谢,增强其致病力。同时,细菌也可能直接损害宿主的肠粘膜,有利于阿米巴的侵袭。
阿米巴致病的免疫机制目前了解得不多。已知阿米巴感染可诱导人和实验动物的体液免疫,产生抗体,但不具有保护和防止再感染的作用。阿米巴病的发病与宿主的细胞免疫状态有关,接受抗淋巴细胞血清注射后的实验动物所发生的病变程度较重。对阿米巴病患者的若干细胞免疫指标测定亦表明有细胞免疫功能的低下。
【病变】
病变部位主要在盲肠、升结肠,其次为乙状结肠和直肠,严重病例整个结肠和小肠下段均可受累。病变为伴组织溶解液化的坏死性炎,可分为急性期和慢性期。
1.急性期病变肉眼观,早期在肠粘膜表面可见多数隆起的灰黄色帽针头大小的点状坏死或浅在溃疡。病变进展时,坏死灶增大,呈圆形钮扣状,周围有出血带包绕。此时滋养体在肠粘膜层内不断繁殖,破坏组织,并穿过粘膜肌层达到粘膜下层。由于粘膜下层组织疏松,阿米巴易于向四周蔓延,坏死组织液化脱落后,形成口小底大的烧瓶状溃疡,边缘呈潜行性(undermined)(图19-1,图19-2),对本病具有诊断意义。溃疡间粘膜正常或仅表现轻度卡他性炎症。在严重的病例,邻近溃疡可在粘膜下层形成遂道样互相沟通,其表面粘膜可大块坏死脱落,形成边缘潜行的巨大溃疡,其直径可达8~12cm。
图19-1 结肠急性阿米巴痢疾
肠粘膜上可见多数大小不等的坏死灶及潜行性溃疡
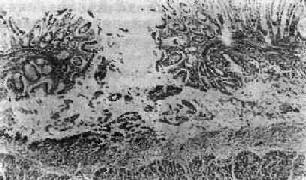
图19-2 急性阿米巴痢疾的结肠溃疡
溃疡深达粘膜下层,溃疡口小底大呈烧瓶状,溃疡口周围的粘膜悬覆于溃疡面上
镜下,阿米巴引起的液化性坏死为无结构的淡红染病灶,其附近组织炎症反应轻微,仅见充血、出血及少量淋巴细胞和浆细胞浸润。溃疡形成时,在溃疡底和边缘仍见残留的坏死组织,周围炎症反应也不明显。如继发细菌感染则可有中性粒细胞浸润。在坏死组织与活组织的交界处常可找到阿米巴滋养体,在肠壁的小静脉腔内有时也可见到(图19-3)。在组织切片上,滋养体一般呈圆形,直径约20~40μm,核小而圆,隐约可见;胞浆略呈嗜碱性,其中可见红细胞、淋巴细胞和组织碎片等。在滋养体周围常有一空隙,可能因组织被溶解所致。
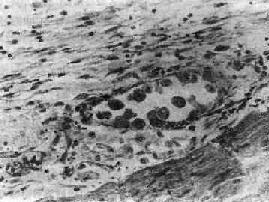
图19-3 急性阿米巴痢疾
结肠粘膜下层小静脉内可见成堆的阿米巴大滋养体
临床上,急性期主要为肠道症状,表现为腹痛、腹泻、大便量增多因含粘液和大量血液及坏死溶解的肠壁组织而呈紫红或暗红色的糊状,伴腥臭。粪检时易找到阿米巴滋养体。由于本病的直肠及肛门病变较轻,故里急后重症状不如细菌性痢疾明显,全身中毒表现也很轻微。
急性期多数可治愈。少数情况下,因溃疡过深,可引起肠穿孔。但因本病病变发展较缓,在穿孔前溃疡底的浆膜层常与邻近组织粘连,故穿孔时仅形成局限性脓肿,很少引起弥漫性腹膜炎。肠壁的小血管破裂引起出血者比较常见,但大血管被破坏导致大出血者则很少见。少数因治疗不够及时、彻底而转入慢性期。
2.慢性期病变病变甚为复杂。一些溃疡已愈合,而另一些溃疡可继续存在并扩大,甚至已愈合的溃疡又再发生坏死。坏死、溃疡、肉芽组织增生和瘢痕形成同时并存,粘膜可增生形成息肉,最终可使肠粘膜完全失去正常形态。肠壁可因纤维组织增生而增厚变硬,甚至引起肠腔狭窄。有时可因肉芽组织增生过多,而形成局限性包块,称为阿米巴肿(amoeboma),多见于盲肠,临床上易误诊为结肠癌。
二、肠外阿米巴病
肠外阿米巴病(extraintestinal amoebiasis)可见于许多器官,以肝、肺及脑为常见。
1.阿米巴肝脓肿阿米巴肝脓肿是肠外阿米巴病中最常见者。国内临床资料统计,阿米巴痢疾合并阿米巴肝脓肿者为1.8%~10%,而尸检统计则为36.6%,甚至高达60%。肝脓肿大多发生于阿米巴痢疾发病后1~3个月内,但也可发生于痢疾症状消失数年之后。阿米巴滋养体系通过侵入肠壁小静脉,经肠系膜静脉、门静脉而到达肝。阿米巴肝脓肿可为单个或多个,但以单个者为多见,且多位于肝右叶(80%)。因原因可能是由于肠阿米巴病多位于盲肠及升结肠,其血液流入肠系膜上静脉,经粗短的门静脉时血流快,来不及与肠系膜下静脉流入的血液相混合而大部分进入肝右叶。此外,肝右叶体积远比左叶为大,故受侵犯的机会也较多。
肉眼观,脓肿大小不等,大者可达小儿头大,几乎占据整个肝右叶。阿米巴肝脓肿的内容非一般脓液,而为阿米巴溶解组织所致的液化性坏死物质和陈旧性血液混合而成的果酱样物质,炎症反应不明显,但习惯上仍称为脓肿。脓肿壁上附有尚未彻底液化坏死的汇管区结缔组织、血管和胆管等,呈破絮状外观(图19-4),具有一定的特征性。
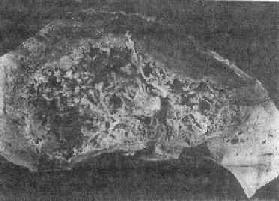
图19-4 阿米巴性肝脓肿
肝右叶为一巨大的脓肿所占据,脓肿壁呈破絮状外观
镜下,见脓肿壁有不等量尚未彻底液化坏死的组织,有少许炎性细胞浸润,在坏死组织边缘的活组织中可查见阿米巴滋养体。慢性脓肿周围可有肉芽组织及纤维组织包绕。
临床上,阿米巴性肝脓肿常表现长期发热伴有右上腹痛及肝肿大和压痛,全身消耗等症状。
阿米巴性肝脓肿可继续扩大并向周围组织穿破。肝右叶脓肿向上穿破时,可在肝和横膈之间形成膈下脓肿。如果肝和膈肌先有粘连,则肝脓肿常破入胸腔、肺,形成脓胸或肺脓肿。阿米巴肺脓肿继而穿破支气管,造成肝-支气管瘘或胸膜-支气管瘘。肝左叶脓肿如向上穿破,可破入纵隔、左胸腔和心包。肝脓肿向下穿破时,可穿入腹腔及腹腔器官,如胃、肠及胆囊等,引起相应部位的阿米巴性炎症。慢性阿米巴性脓肿常继发细菌感染而与一般细菌引起的脓肿相似,其脓液呈黄色或黄绿色,病情也相应恶化。
2.阿米巴性肺脓肿很少见,绝大多数是由肝脓肿穿过横膈直接蔓延而来。脓肿常位于右肺下叶,为单发性,由于横膈被穿破,故肺脓肿常与肝脓肿互相连通。脓肿腔内含咖啡色坏死液化物质,如破入支气管,坏死物质被排出后形成空洞。临床上患者有类似肺结核症状,咳出褐色脓样痰,其中可检见大量阿米巴滋养体。
3.阿米巴性脑脓肿极少见,往往是肝或肺脓肿内的阿米巴滋养体经血道进入脑而引起。
偶尔直肠的阿米巴病变可直接蔓延至肛周和会阴皮肤,引起边缘也为潜行性的溃疡。阴道、宫颈、精囊腺、前列腺、尿道等器官亦可被侵犯。
第二节 血吸虫病
血吸虫病(schistosomiasis)是由血吸虫寄生于人体引起的地方性寄生虫病。寄生于人体的血吸虫主要有三种:即流行于非洲北部的埃及血吸虫(Schistosoma haematobium);流行于拉丁美洲及非洲中部的曼氏血吸虫(Schistosoma mansoni)以及流行于亚洲的日本血吸虫(Schistosoma japanicum)。在我国因只有日本血吸虫病流行,故通常将日本血吸虫病简称为血吸虫病。
根据1972年在湖南长沙马王堆一号墓出土的西汉女尸及1975年在湖北江陵出土的西汉男尸内皆发现有大量典型血吸虫卵的事实,证明在2100多年前我国已有血吸虫病的流行。本病在我国主要流行于长江流域及其以南的十三个省市的广大水稻作物地区,尤以江苏、浙江、安徽、湖北、湖南、江西等省及上海市效较为严重。过去本病流行猖獗,对广大劳动人民的健康危害极大。新中国成立后,积极发展了防治工作,广东、广西、福建、江苏和上海等五个省市和全国270多个县已基本消灭了血吸虫病。
【病因及感染途径】
日本血吸虫的生活史可分为虫卵、毛蚴、胞蚴、尾蚴、童虫及成虫等阶段。成虫以人体或其他哺乳动物如狗、猫、猪、牛及马等为终宿主,自毛蚴至尾蚴的发育繁殖阶段以钉螺为中间宿主。
血吸虫虫卵随同病人或病畜的粪便排入水中,卵内的毛蚴成熟孵化,破壳而出,以后钻入钉螺体内,经过母胞蚴及子胞蚴阶段后,大量尾蚴发育成熟,并游动于水中。当人畜与疫水接触时,尾蚴借其头腺分泌的溶组织酶作用和其肌肉收缩的机械运动,很快钻入皮肤(或粘膜)并脱去尾部变为童虫。童虫经小静脉或淋巴管进入血液循环,再经右心而到达肺。以后由肺的毛细血管经肺静脉而入大循环向全身散布。只有进入肠系膜静脉的童虫,才能继续发育为成虫,其余多在途中夭折。通常在感染尾蚴后3周左右即可发育为成虫,雌雄成虫交配后即可产卵。虫卵随门静脉血流顺流到肝,或逆流入肠壁而沉着在组织内,约经11天左右逐渐发育为成熟虫卵,内含毛蚴。肠壁内的虫卵可破坏肠粘膜而进入肠腔,并随粪便排出体外,再重演生活周期。虫卵在组织内的寿命约为21天左右。雌雄合抱的成虫在人体内的寿命一般为3~4年。
【病变和发病机制】
血吸虫发育阶段中的尾蚴、童虫及成虫、虫卵等均可引起病变,但以虫卵引起的病变最严重,危害也最大。
1.尾蚴及童虫引起的病变
(1)尾蚴性皮炎:尾蚴侵入皮肤后,可引起皮肤的炎症反应,称为尾蚴性皮炎(cercarial dermatitis)。多发生于重复感染的患者,一般在尾蚴钻入皮肤后数小时至2~3日内发生,表现为红色小丘疹,奇痒,经数日后可自然消退。镜下见真皮充血、出血及水肿,起初有中性及嗜酸性粒细胞浸润,以后主要为密集的单核细胞浸润。
目前认为本病的发病机制主要与迟发性变态反应有关。动物实验证明,转移感染小鼠的淋巴细胞给正常小鼠,再接种尾蚴,经1~2天,局部童虫周围有单核细胞浸润。在反应早期可能有抗体介导的免疫反应参与。
(2)肺部病变:童虫移行到肺时,部分可穿破肺泡壁毛细血管,游出到肺组织中,引起点状出血及白细胞浸润(约在感染后1~2天)并可有血管炎改变,但病变一般轻微而短暂。
童虫经大循环移行到其他器官时也可引起与肺类似的改变。
童虫所引起的各器官点状出血除与童虫的机械作用有关外,还与其代谢产物或虫体死亡后蛋白分解产物所致人体组织的变态反应有关。
宿主感染血吸虫后得到获得性免疫,对再感染产生不同程度的抵抗力。其机制为抗体依赖、细胞介导的细胞毒反应,由IgE或IgG2a抗体,巨噬细胞、嗜酸性粒细胞等参与。主要作用于表面有抗原表达的幼龄童虫,嗜酸性粒细胞有IgG和IgE的Fc受体,当抗体包被童虫后其Fc段与Fc受体结合,并使嗜酸性粒细胞粘附在童虫表面且脱颗粒,释出细胞毒性物质,而起杀伤作用。巨噬细胞也是非常重要的效应细胞。
2.成虫引起的病变血吸虫在门静脉系统内发育成熟后,其代谢产物可使机体发生贫血、嗜酸性粒细胞增多、脾肿大、静脉内膜炎及静脉周围炎等。在肝、脾的单核吞噬细胞系统的细胞内,常见有黑褐色血吸虫色素沉着,是成虫吞食红细胞后,在虫体内珠蛋白酶作用下,使血红蛋白分解而形成的一种血红素样色素,同样的色素也见于成虫的肠道内。活的成虫本身在静脉内不引起宿主反应,其解释是成虫的表膜内含有宿主的抗原,被宿主认为是“自我”组织而逃避了免疫攻击。成虫死亡后,多在肝内分解,产生毒性,可引起明显的静脉炎和静脉周围炎。死亡虫体周围组织坏死,大量嗜酸性粒细胞浸润,形成嗜酸性脓肿,与血丝虫成虫死亡后引起的变化相似。
3.虫卵引起的变化虫卵沉着所引起的损害是最主要的病变,虫卵除主要沉着于乙状结肠和直肠壁以及肝外,也常见于回肠末段、阑尾及升结肠等处。肺、脑等其他器官有时也可见到。未成熟的虫卵所引起的病变轻微;含毛蚴的成熟虫卵往往引起虫卵结节形成。按其病变发展过程可分为急性虫卵结节和慢性虫卵结节两种。
(1)急性虫卵结节:肉眼观为灰黄色、粟粒至绿豆大(0.5~4mm)的小结节。镜下见结节中央常有1~2个成熟虫卵,也偶可多达20个以上。这些成熟虫卵的卵壳上附有放射状嗜酸性的棒状体,也称为Hoeppli现象,用免疫荧光法已证实为抗原抗体复合物。虫卵周围是一片无结构的颗粒状坏死物质及大量嗜酸性粒细胞浸润。因其病变类似脓肿,故也称为嗜酸性脓肿(图19-5)。在坏死组织中可混杂多数菱形或多面形屈光性蛋白质晶体,即Charcot-Leyden结晶,系嗜酸性粒细胞的嗜酸性颗粒互相融合而成。随后虫卵周围产生肉芽组织层,其中有以嗜酸性粒细胞为主的炎症细胞浸润,还有单核巨噬细胞、淋巴细胞、浆细胞及少量中性粒细胞。随着病程的发展,肉芽组织层逐渐向虫卵结节中央生长,并出现围绕结节呈放射状排列的类上皮细胞层。类上皮细胞层逐渐加宽,嗜酸性粒细胞显著减少,构成晚期急性虫卵结节(图19-6),这是向慢性虫卵结节发展的过渡阶段。
图19-5 肝血吸虫病之急性虫卵结节
结节中心有一成熟虫卵,卵壳表面可见放射状物质,周围广泛坏死伴大量嗜酸性粒细胞浸润
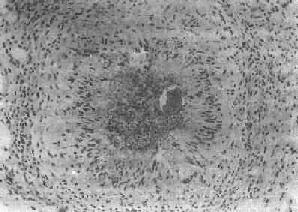
图19-6 血吸虫病之晚期急性虫卵结节
坏死区及嗜酸性粒细胞浸润范围缩小,其周围出现放射状排列的类上皮细胞层
(2)慢性虫卵结节:急性虫卵结节经10余天后,虫卵内毛蚴死亡,坏死物质逐渐被吸收,虫卵破裂或钙化,其周围除类上皮细胞外,出现异物巨细胞和淋巴细胞,形态上似结核结节,故称为假结核结节(图19-7)。少数虫卵结节一开始即为假结核结节,而不经过急性虫卵结节阶段。最后,假结核结节中的类上皮细胞为纤维母细胞代替,并产生胶原纤维,使结节纤维化。其中央的卵壳碎片及钙化的死卵可长期存留。
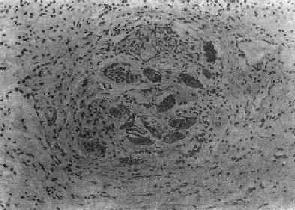
图19-7 血吸虫病之慢性虫卵结节
结节中央有破裂和钙化的虫卵,其周围有类上皮细胞和异物巨细胞,形成假结核结节
目前认为虫卵结节的形成主要是由T细胞介导的迟发性变态反应的结果。诱发虫卵结节的抗原来自成熟虫卵中毛蚴所分泌的可溶性卵抗原(soluble egg antigen,SEA),是一种含蛋白质、糖蛋白、多糖和酶的异质性混合物,其糖蛋白中的某些成分,如MSA,gp-2是引起虫卵肉芽肿的主要抗原。活毛蚴分泌的抗原,透过卵壳上的微孔,持续稳定地释出,使机体致敏。经巨噬细胞吞噬、加工的抗原,传递给辅助性T细胞(TH),致敏的TH产生多种淋巴因子,如巨噬细胞激活因子(MAF)、嗜酸性粒细胞趋化因子(ECF)等,引起巨噬细胞和嗜酸性粒细胞等聚集于虫卵周围而形成虫卵肉芽肿。实验证明淋巴细胞或抗巨噬细胞的血清、细胞免疫抑制药物等均能抑制致敏小鼠虫卵肉芽肿的形成。新生期摘除胸腺的小鼠和先天性无胸腺裸鼠体内形成的虫卵肉芽肿均较对照组小。致敏小鼠和感染小鼠的淋巴细胞可将致敏被动地转移给受体小鼠,这些都说明T细胞在虫卵肉芽形成过程中起重要作用。除细胞免疫外,在急性虫卵结节形成过程中,毛蚴分泌的SEA刺激宿主产生抗体,在虫卵周围形成免疫复合物,Ⅲ型变态反应亦可能参与虫卵结节的形成。
这种肉芽肿的形成是宿主对虫卵的一种免疫应答,使虫卵变性、破坏、被吞噬和清除,还可以隔离和中和虫卵释放的抗原及毒性物质,起局部免疫屏障作用。另一方面,在早期肉芽肿内即可测出纤维连结蛋白(FN),体外培养的肉芽肿能分泌纤维母细胞生长因子(FGF),说明虫卵结节能破坏宿主正常组织并导致器官纤维化。
【主要器官的病变及其后果】
1.结肠病变常累及全部结肠,以乙状结肠最为显著。这是因为日本血吸虫成虫多寄生于肠系膜下静脉和痔上静脉的缘故。
早期肉眼观,肠粘膜红肿,呈急性卡他性炎,隐约可见褐色或灰黄色细颗粒状扁平隆起的病灶(虫卵堆积所致),直径约0.5~1cm左右。病灶中央可发生坏死脱落形成浅表溃疡,其边缘常有充血。虫卵可随坏死组织脱落入肠腔,在粪便中可查见虫卵。镜下,见粘膜及粘膜下层有成堆虫卵堆积,形成急性虫卵结节,尤以粘膜下层为明显。溃疡一般较小且表浅,深达粘膜肌层或粘膜下层,如邻近的小溃疡互相融合,可形成较大溃疡。在肠病变的早期,临床可出现腹痛、腹泻等痢疾样症状。
随着病变的发展形成假结核结节,最后发生纤维化,虫卵也逐渐死亡及钙化。由于虫卵的反复沉着,引起肠粘膜反复发生溃疡和肠壁纤维化,最终导致肠壁增厚变硬,甚至肠腔狭窄和肠梗阻。肠粘膜粗糙不平,萎缩,皱襞消失,除见小溃疡外,还可见多发性小息肉(图19-8)。由于肠壁结缔组织增生,使以后到达肠壁的虫卵难于排入肠腔,故晚期患者粪便中不易查见虫卵,一般需做直肠粘膜压片、活检或皮内试验等来确诊本病。

图19-8 慢性血吸虫病的结肠
肠壁增厚,粘膜粗糙不平,多处粘膜呈息肉样增生
肠慢性血吸虫病可并发结肠癌,这类结肠癌的特点是:①发病年龄较轻,平均年龄约比一般结肠癌小8岁;②发生癌变部位和肠道血吸虫病一致,多见于乙状结肠及直肠,该肠段常有严重慢性血吸虫病的改变;③在慢性血吸虫病肠粘膜增生性息肉的基础上有逐渐演变为癌的过渡形态,偶尔可见数个息肉同时发生癌变。
2.肝肉眼观,早期可有轻度肝肿大,表面及切面可见多个不等的灰白或灰黄色、粟粒或绿豆大小的小结节(图19-9)。镜下,见急性虫卵结节,主要分布在汇管区附近,肝细胞可因而受压萎缩,门静脉分支可有静脉内膜炎改变。也可有变性及小灶性坏死。Kupffer细胞内可见黑褐色血吸虫色素沉着。
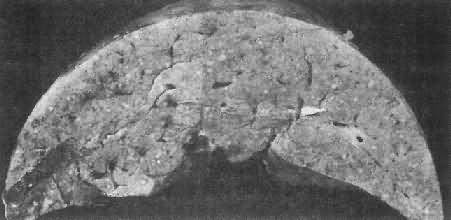
图19-9 肝急性血吸虫病
肝表面及切面上可见多数灰白色急性虫卵结节
慢性病例肝内可见慢性虫卵结节和纤维化。在长期重度感染的病例,肝因严重纤维化而变硬、变小,导致血吸虫性肝硬变。肝表面不平,有浅沟纹构成微隆起的分区,严重者可形成粗大突起的结节(图19-10)。切面上,增生的结缔组织沿门静脉分支呈树枝状分布,故称为干线型或管道型肝硬变(pipe stem cirrhosis)(图19-11)。较大门静脉分支管壁增厚,并可有血栓形成(血栓性静脉炎)。由于虫卵结节主要见于汇管区,肝小叶并未遭受严重破坏,故不形成假小叶,与门脉性肝硬变不同。由于门静脉分支虫卵栓塞、静脉内膜炎、血栓形成和机化,以及门静脉周围纤维组织增生,使肝内门静脉分支阻塞和受压,从而造成门静脉高压。因肝内门静脉的阻塞是窦前性的,故门静脉高压较门脉性肝硬变时更为显著,临床上常出现腹水、巨脾、食管静脉曲张等后果。
图19-10 血吸虫病性肝硬变
肝表面呈粗大结节状
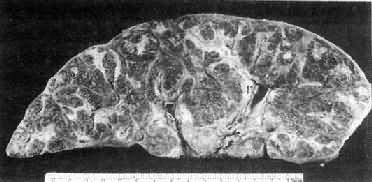
图19-11 血吸虫病性肝硬变
肝切面见沿门静脉分支有大量的纤维组织增生
3.脾早期肿大不明显,主要由于成虫的代谢产物引起的单核巨噬细胞增生所致。晚期主要由门静脉高压引起的脾淤血所致,此时可形成巨脾,重量可达1000g,甚至可达4000g。肉眼观,脾质地坚韧,包膜增厚。切面呈暗红色,脾小梁清楚,脾小体多不明显,常见棕黄色的含铁小结,有时还可见多数梗死灶。镜下,脾窦扩张充血,窦内皮细胞及网状细胞增生,窦壁纤维组织增生而变宽。脾小体萎缩减少,单核巨噬细胞内可见血吸虫色素沉着。陈旧出血灶伴有铁质及钙盐沉着和纤维组织增生,形成含铁小结(siderotic nodule)。脾内偶见虫卵结节。临床上可出现贫血、白细胞减少和血小板减少等脾功能亢进症状。
4.肺在部分急性病例,肺内可出现多数急性虫卵结节,其周围肺泡出现炎性渗出物,X线照片类似肺的粟粒性结核。通常肺的变化甚轻微,一般不导致严重后果。关于肺内虫卵的来源,近年来认为并非成虫寄生在肺内产卵,而主要是通过门-腔静脉之间的交通枝而来。在肝内门静脉分支严重阻塞并发门静脉高压的患者,更易发生门-腔静脉交通枝的开放。
5.其他器官脑的血吸虫病主要见于大脑顶叶,也可累及额叶及枕叶,表现为不同时期的虫卵结节形成和胶质细胞增生。临床上出现脑炎、癫痫发作和疑似脑内肿瘤的占位性症状。关于虫卵进入脑的途径,最大可能是肺部虫卵经肺静脉到左心,而后由动脉血流带入脑内。近年来发现由血吸虫感染引起的血吸虫病肾小球肾炎,肾小球内发现有IgG及补体C3的沉着,故属于Ⅲ型变态反应引起的免疫复合物肾炎。在严重感染的病例中,胰腺、胆囊、心、肾、膀胱及子宫等器官内也可见虫卵沉积,但数量少,组织反应一般不甚明显。
附 急性血吸虫病
多见于初次感染大量尾蚴后1个月左右,成虫开始大量排卵时。临床表现为发病急骤,全身中毒反应显着,有发热,血中嗜酸性粒细胞及免疫球蛋白增高,肝、脾及全身淋巴结肿大,肝区触痛,并有腹泻或痢疾症状及全身荨麻疹等。病情严重者,可引起死亡。受累器官主要是肝和结肠,但肺、脑及其他器官也可受累。病变以浆液性炎和出血性炎以及急性虫卵结节形成为主,脾和淋巴结可有淤血、单核巨噬细胞增多以及嗜酸性粒细胞和浆细胞浸润。实质性器官如心、肝、肾的变性和血管炎改变有时也很显着。
急性血吸虫病的发病机制尚不清楚,目前研究认为可能是一种免疫复合物病或血清病。在血吸虫感染的早期,尾蚴和移行的童虫可刺激宿主产生抗体,但抗体的水平较低。当童虫发育为成熟成虫并大量产卵时,虫卵释放出来的大量虫卵可溶性抗原,刺激宿主迅速产生抗体,在抗原过剩的情况下,形成可溶性抗原抗体复合物造成血管损害而致病。当新的抗体迅速形成并超过抗原含量或当虫卵周围肉芽组织形成,将抗原隔离时,则病情可终止。
第三节 华支睾吸虫病
华支睾吸虫病(clonorchiasis sinensis)是由华支睾吸虫(clonorchis sinensis)成虫寄居引起,因其主要寄生在肝内胆管,故俗称为肝吸虫病。本病流行于印度、越南、菲律宾等东南亚国家和中国、日本、朝鲜等国。在我国,广东及台湾为主要流行区,但北方各省也有流行区或散发的报告。根据1975年湖北江陵西汉古尸及1994年荆门战国古尸研究,在死者肠内容物内均发现本虫虫卵,说明本病在我国存在至少已有2300多年历史。
【病因及感染途径】
含有成熟毛蚴的虫卵随人或狗、猫和猪等终宿主的粪便入水后,可被第一中间宿主淡水螺吞食,在其消化道内,虫卵内的毛蚴顶开卵盖脱壳而出,并逐渐发育为尾蚴。尾蚴自螺体逸出后在水中游动,遇到第二中间宿主淡水鱼或淡水虾时,在其体内,尤其是肌肉内发育为囊蚴。
当人或动物食入未经煮熟的含活囊蚴的鱼或虾后,囊蚴经胃肠消化酶及胆汁的作用,在十二指肠内脱囊而发育成童虫。童虫经总胆管至肝内胆管寄生并发育为成虫。从食入囊蚴至粪便中出现虫卵约需1个月。
【病变及合并症】
被成虫寄生的肝内胆管,其病变程度因感染轻重而异,轻者感染虫数十余条至数十条,无明显肉眼病变。重度感染常达数千条以上,充满肝内、外胆管,在胆总管、胆囊及胰导管内也可见到。病变的发生除与虫体的机械性阻塞及代谢或崩解产物的化学刺激有关外,虫体产生的抗原所引起的过敏反应也起一定作用。
1.肝肉眼观,肝轻度肿大,尤以左叶为著,严重时在左叶被膜下即可见到因成虫机械阻塞而扩张的胆管分支,可能因左叶胆管较平直,易被童虫侵入之故。切面见肝内大、中胆管呈不同程度扩张和管壁增厚,胆管的直径可达3~6mm,壁可厚达0.5~3mm(图19-12)。胆管腔内充满胆汁,并含有数目不等的成虫。华支睾吸虫前端较细,后端钝圆,形似葵瓜子,虫体长约10~25mm,宽约3~5mm,柔软而半透明。在有大量虫体寄生的病例,解剖时轻压肝脏,即可见成虫由各胆管中鱼贯而出。镜下,根据感染虫数和感染持续时间的不同,病变可轻重不等。肝内胆管扩和,胆管上皮细胞呈不同程度增生,严重者上皮向管腔内呈乳头状增生,并可在粘膜下见多量的增生腺体,形成腺瘤样结构(图19-13)。上皮细胞还常发生杯状细胞化生而分泌大量粘液。管壁有不等量淋巴细胞、浆细胞和嗜酸性粒细胞浸润。慢性病例则伴有明显的纤维结缔组织增生。部分病例汇管区的结缔组织也呈轻度增生,伴有上述炎性细胞浸润。在急性反应时汇管区的小胆管周围也可有多量嗜酸性粒细胞浸润,而肝实质细胞一般无明显改变。
图19-12 肝华支睾吸虫病
切面见大、中胆管扩张,管壁增厚
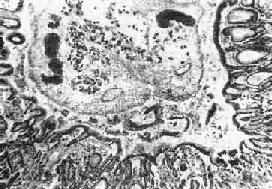
图19-13 肝华支睾吸虫病
胆管内可见虫体,胆管粘膜有腺瘤样增生及炎症
成虫在肝内胆管寄生,使胆管内胆汁淤积,容易发生继发感染。死亡的虫体、虫卵和脱落的胆管上皮还可以成为胆石的核心,促进胆石形成。
关于华支睾吸虫感染与肝癌(胆管上皮癌)的关系,我国病理学者侯宝璋、秦光煜等曾分别对人体或自然感染的动物(家猫及家犬)进行了观察研究,证实在华支睾吸虫感染的致的胆管上皮腺瘤样增生的基础上,可以发生癌变,在人工感染的家猫实验中也得到证实。至于致癌因素是成虫在胆管内蠕动的机械性刺激或虫体代谢产物和胆汁成分的化学刺激,尚有待进一步研究。
2.胆囊成虫也可在胆囊内寄生(约占9%),镜下可见胆囊壁有嗜酸性粒细胞及淋巴细胞浸润,而上皮细胞增生多不明显。
3.胰腺成虫也可在胰腺导管内寄生(占6%~37.5%)引起病变,可能是成虫随胆汁进入胰管,也可能是童虫进入胰管内发育为成虫所致。肉眼观,胰管扩张、增厚。镜下,可见胰管上皮增生,并伴有不同程度的鳞状化生(可达95%)。鳞状化生的程度往往与感染的虫数有关。胰腺实质一般无明显改变,但有报告谓可诱发急性胰腺炎。
第四节 肺吸虫病
肺吸虫病(paragonimiasis)主要是肺吸虫虫体(童虫及成虫)在人体内穿行或寄居所引起的疾病,病变以在器官或组织内形成互相沟通的多房性小囊肿为特点。近年来对肺吸虫的研究获得较大的进展,发现了许多新亚种。在我国除东北和浙江老疫区外,还发现安徽、江苏以及四川、贵州、湖北、湖南及河南等省的新流行区。前者以卫氏肺吸虫(Paragonimus westermani)感染为主;后者以斯氏肺吸虫(Paragonimus skrjabini)感染为主。亚洲各国如朝鲜、日本及菲律宾等也有本病流行。
【病因及感染途径】
肺吸虫各虫种的生活史及其与宿主的关系基本相同。成虫主要寄生在人及其他哺乳动物如猫、犬、猪、虎、豹等的肺内,也可寄生于肺以外的组织或器官。卫氏肺吸虫虫体大小约为9×5×3mm,呈扁平卵圆形,具有较强的收缩运动能力。虫卵主要随痰咳出,以后在水内孵化成毛蚴。毛蚴侵入第一中间宿主淡水螺内经过胞蚴、雷蚴等发育阶段最后成为尾蚴。尾蚴进入第二中间宿主淡水石蟹或蝲蛄的体内发育成为囊蚴。人若进食含有此种囊蚴的生的或未煮熟的石蟹或蝲蛄时,囊蚴随之进入消化道,经消化液作用脱囊成为童虫。童虫的活动能力很强,加上所分泌的酶的作用,可穿过肠壁到腹腔浆膜表面匍匐游行,其中多数童虫沿肝表面向上移行,直接贯穿膈而达胸腔,进而侵入肺内并发育为成虫。少数童虫停留于腹腔内,继续发育,并穿入肝脏浅层或大网膜成为成虫。偶尔可沿纵隔内大血管根部及颈内动脉周围软组织向上移行,经破裂孔而侵入颅中凹,再经颞叶、枕叶的底部侵入脑组织。虫体侵入器官或组织后除引起该处病变外,还可以继续穿行到其他部位,引起病变。一般从囊蚴进入体内到在肺内成熟产卵,约需2~3个月。在虫在宿主体内一般可活5~6年。
【病变】
肺吸虫的致病作用,主要由虫体(童虫及成虫)引起。虫体在组织内游走或定居,对局部组织造成机械性损伤;虫体代谢产物等抗原物质会导致人体的免疫病理反应。由于肺吸虫卵在人体内不能发育成毛蚴,不分泌可溶性抗原,因此与血吸虫卵所致的病变不同,仅引起异物肉芽肿反应。
【基本病变】
(1)浆膜炎:虫体在体腔内移行和寄生时,在早期引起纤维素性或浆液纤维素性腹膜炎或胸膜炎,渗出液内可找到虫卵,具有临床诊断价值。日久后可因纤维化引起腹腔内器官间粘连、胸膜粘连甚至胸腔闭锁。
(2)组织破坏及窦道形成:虫体在组织中穿行时引起坏死及出血,形成迂迴的窟穴状病灶或窦道(图19-14),其中可见虫卵,周围有轻度嗜酸性粒细胞及淋巴细胞浸润,以后可发展为纤维化。
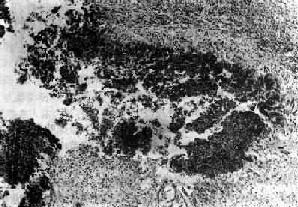
图19-14 肺吸虫病的窦道
在嗜酸性白细胞浸润的基础上,有坏死腔穴和窦道形成,虫体已移行他处
(3)虫囊肿形成:童虫或成虫在器官内定居时,最初引起组织坏死及出血,继而引起强烈的炎性反应,其中除嗜酸性粒细胞外,还有多量中性粒细胞而形成脓肿。随后脓肿周围有肉芽组织及纤维组织包绕,形成囊肿。囊内可含有虫体、虫卵及Charcot-Leyden结晶,囊内容物为棕色粘稠液体。进入囊壁内和周围组织的虫卵,可形成异物肉芽肿。由于虫体有游走习性,故可离开原囊肿,在其附近继续破坏组织,形成新囊肿。囊肿间常以窦道互相沟通,形成多房性囊肿。囊肿内成虫如迁移或死亡、溶解后,囊内容物可被逐渐吸收,囊肿为肉芽组织所充填,最后形成瘢痕。
【各器官的病变】
(1)肺:胸膜广泛粘连,在增厚的胸膜和肺内有新旧不一、散在或群集的虫囊肿,囊肿大小不等,囊内可找到虫体和虫卵(图19-15)。肺的虫囊肿常侵犯支气管壁,使之与囊肿相通。临床上患者每因上述病变而有胸痛、咳嗽、痰中带血或烂桃样血痰。痰中可查见虫卵、嗜酸性粒细胞和Charcot-Leyden结晶。囊肿及其周围肺组织可继发细菌感染,有时可并发气胸、脓胸甚至血胸。慢性病例有明显的肺纤维化。
图19-15 实验性狗肺吸虫囊肿之剖面
囊内可见两条肺吸虫
(2)腹腔:腹腔器官间往往有广泛粘连。在大网膜、肠系膜和大、小肠表面,甚至肝表面,可见分散或成群的虫囊肿。囊肿多者可形成肿块。
(3)脑:脑的受累以儿童及青年较多。病变多见于大脑颞叶及枕叶,侵犯小脑者少见。由于脑组织柔软,虫体易于移行,故可侵入内囊、基底节、顶叶、额叶、脑室及对侧大脑。虫体穿过脑室时,常可在脑脊液内查见虫卵。脑的肺吸虫病病变与肺部所见大致相同,虫囊肿周围组织可有出血、软化及胶质细胞增生。
(4)皮下结节:系成虫从腹腔穿入软组织所致,比较少见,约见于10%的病例。皮下结节可呈游走性(因虫体移行所致),并主要分布于腹、背、臀、阴囊及大腿等处,由虫囊肿构成,大小约为1.5~2.5cm,可成群、成串出现,如穿通皮肤则可形成溃疡。
(5)其他器官:腹腔内的虫体如穿行到腹膜后组织,可累及肾周脂肪组织、肾、肾上腺、腰大肌等。虫体如穿入背部深层肌肉,甚至穿过胸椎间孔(大多在第10胸椎水平上下)而进入硬脊膜腔,可在该处形成虫囊肿而压迫脊髓。虫体如穿过腹股沟内环,可引起精索和阴囊受累。此外,眼窝、眼睑、心包等处亦有受累的报告。
近年来在四川、云南、贵州、湖南等地发现一种肺吸虫病,大多认为系斯氏肺吸虫所致,但亦有认为是由一种新型肺吸虫——四川肺吸虫引起者。与卫氏肺吸虫不同,人体不是它的适宜宿主,虫体侵入人体后不易发育为成虫和产卵,故组织中所见多为童虫。它引起的全身和局部过敏反应均较强烈,血中嗜酸性粒细胞明显增高,局部组织炎症反应中亦以嗜酸性粒细胞为主,而中性粒细胞较少。肺部损害较轻,而以多发性、游走性皮下结节为多见。主要分布于胸、腹部及大腿内侧。皮下结节内没有虫卵,也很难查见虫体偶可见到童虫。由于虫体进入肝的数目较多,而且在肝内寄居的时间较长,因而可引起明显的甚至严重的肝损害。脑内出血并伴蛛网膜下腔出血者也较多见。其他如眼眶、心包、胸膜等亦可受累。
第五节 丝虫病
丝虫病(filariasis)是由丝虫寄生于人体淋巴系统所引起的疾病。本病以蚊为传播媒介,早期以淋巴管炎及淋巴结炎为主,晚期则以淋巴回流障碍为主,出现淋巴管扩张及象皮肿等。本病流行于世界各地,发热带及亚热带地区为多见。在我国流行甚广,如山东、河南、江苏、浙江、福建、台湾、广东、广西、湖南及湖北等省区均有流行。
【病因和感染途径】
目前已知寄生在人体的丝虫共8种,但在我国流行的只有班氏丝虫(Wuchereria bancrofti)和马来丝虫(brugia malayi)两种,前者主要由库蚊传播;后者由中华按蚊传播。两者生活史基本相似。
当蚊叮人吸血时,蚊体内的感染期幼虫钻入人体。一般认为幼虫迅速侵入附近的淋巴管,并移行至大淋巴管及淋巴结寄生,发育为成虫。马来丝虫主要寄生在上、下肢的浅表淋巴系统,尤以下肢为多;班氏丝虫除寄生在浅表淋巴系统外,多寄生于深部淋巴系统中,如下肢、阴囊、精索、肾盂等部位。当雌雄虫体交配后,雌虫即产生微丝蚴。微丝蚴自淋巴系统进入血液循环,一般白天滞留于肺及其他器官的毛细血管内,夜间开始出现于周围血液中。这种夜现周期性机制尚未完全阐明,可能与宿主的生理状况、生活习惯,尤其是睡眠习惯有关;亦与微丝蚴特有的生物节律有关,这种节律受宿主的昼夜节律影响并与之同步。丝虫从感染期幼虫侵入人体至发育为成虫并产生微丝蚴的时间,一般约需8~12个月。微丝蚴在人体内可存活2~3个月,成虫约可存活3年。
【发病机制】
对丝虫病的发病机制至今尚未完全阐明,丝虫病的发生与发展取决于多种因素,与宿主的机体反应性、感染的虫种、程度和次数以及虫体的发育阶段、寄居部位和成活情况等因素有关。一些实验证明,丝虫的感染期幼虫、成虫和微丝蚴以及其代谢产物都具有抗原性,机体可产生对抗丝虫的特异性抗体。人体感染丝虫后,血清中IgG和IgE水平均有升高。实验还证明,丝虫感染后除产生体液免疫外,还可能有细胞免疫参与,例如实验发现致敏动物还可出现皮肤的迟发型变态反应和巨噬细胞移动抑制现象。此外还观察到切除胸腺的小鼠对丝虫的易感性增高,并出现微丝蚴血症。一般认为,在丝虫病的急性期变态反应起重要作用。童虫和成虫的代谢产物,尤其是感染期幼虫蜕皮时的分泌物,雌性成虫子宫分泌物以及死虫及其分解产物均可引起局部和全身的变态反应。晚期丝虫病与丝虫成虫阻塞淋巴流有重要关系。但晚期患者发生进行性象皮肿时,常不能证明宿主体内还有活丝虫存在,血中也难以查见微丝蚴。患者血清中IgG升高,因此在晚期丝虫病发病机制中是否还有自身免疫因素存在,尚待证实。人体对丝虫感染的获得性免疫既不能彻底消除已感染的虫体,也不能防止再感染。
【病变】
丝虫的微丝蚴和成虫均可引起病变,但对人体造成严重危害者是成虫所致的病变。
1.微丝蚴所致病变微丝蚴以肺内为最多,心肌及肾次之。一般不引起明显病变。偶尔在脾、脑及乳腺可引起微丝蚴肉芽肿,呈结核样结节,伴有较多的嗜酸性粒细胞浸润。当微丝蚴死亡、钙化后,可引起异物巨细胞反应及纤维结缔组织增生。
2.成虫所致病变主要引起淋巴结及淋巴管的病变,活虫引起的反应一般较轻,而死虫每引起剧烈的组织反应。病变可分为急性期及慢性期。
(1)淋巴管炎:多发生在较大的淋巴管,以下肢、精索、附睾、腹腔内淋巴管及乳腺等处较多见。肉眼观,急性期发炎的淋巴管呈一条红线样自上而下蔓延,形成所谓离心性淋巴管炎。当皮肤表浅微细淋巴管亦被波及时,局部皮肤则呈弥漫性红肿,称为丹毒性皮炎。镜下,常见淋巴管扩张、内皮细胞肿胀增生,管壁水肿增厚和嗜酸性粒细胞及单核细胞浸润。虫体死亡后对组织刺激强烈,引起凝固性坏死及大量嗜酸性粒细胞浸润,形成所谓嗜酸性脓肿。坏死组织中央可见死亡虫体断片及脱出在虫体外的微丝蚴,病变附近可找到Charcot-Leyden结晶。慢性期在脓肿周围出现类上皮细胞、巨噬细胞及异物巨细胞或Langhans巨细胞,形成结核样肉芽肿(图19-16)。随着虫体的钙化,肉芽肿逐渐纤维化,形成同心圆状排列的实心纤维索,使管腔完全闭塞,形成闭塞性淋巴管炎,而引起一系列继发改变。
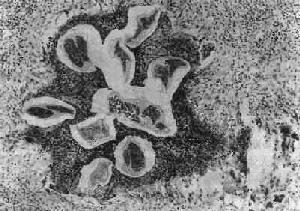
图19-16 丝虫性淋巴管炎
图中可见数个切断的死虫体,周围有嗜酸性脓肿及肉芽肿反应
(2)淋巴结炎:一般由成虫寄居于淋巴结引起,较多见于腹股沟、腘窝及腋窝等处淋巴结。肉眼观,淋巴结显著肿大。镜下,见病变的发展过程与上述淋巴管炎的改变基本相同。死亡虫体钙化后,病变可逐渐纤维化成为瘢痕,影响淋巴液的流通,而导致淋巴淤滞。
(3)淋巴系统阻塞引起的病变:长期反复感染的丝虫性淋巴管炎和淋巴结炎可引起淋巴系统的回流障碍,从而发生一系列改变。
1)淋巴窦及淋巴管扩张:淋巴结内的淋巴窦扩张,形成局部囊状肿块,常见于腹股沟淋巴结,也称腹股沟淋巴结曲张(varicose groin gland),穿刺淋巴液,其中可找到微丝蚴。阻塞远端的淋巴管可见淋巴淤滞而引起曲张,常见于精索、阴囊及大腿内侧,造成组织水肿。淋巴管极度曲张时可使管壁破裂和淋巴液外溢。根据淋巴系统阻塞的部位不同,所溢出的淋巴液性质和病变影响也各异。
当阻塞发生在肠干淋巴管入口的上方或主动脉前淋巴结时,因乳糜液不能回流至乳糜池,则胸导管以下的远端淋巴管皆发生曲张,并形成侧支循环与主动脉侧淋巴结相通。此时如乳糜液经曲张的侧支循环返流至肾盂、输尿管或膀胱的淋巴管而引起破裂时,乳糜液乃溢入尿中,形成乳糜尿。如果乳糜液由此至精索淋巴管流入睾丸鞘膜内,则引起鞘膜乳糜积液;乳糜液也可通过肠系膜淋巴管进入腹腔,则形成乳糜腹水。当阻塞部位发生在肠干淋巴管入口处下方的腰干淋巴管或主动脉侧淋巴结等处时,则对乳糜液回流到胸导管无影响。此时淤滞的淋巴液逆流,则引起相应的淋巴尿、淋巴腹水和鞘膜淋巴积液。
2)象皮肿(elephatiasis):是晚期丝虫病的最突出病变。病变皮肤及皮下组织明显增厚、粗糙、肥大而下垂,皮皱加深,有如大象的皮肤外观,因而得名。有时尚可伴有苔藓样变、棘刺及疣状突起等变化。镜下,表皮角化过度和棘细胞肥厚,真皮及皮下有致密纤维组织极度增生,弹力纤维消失,淋巴管和小血管周围有少许淋巴细胞、浆细胞及嗜酸性粒细胞浸润。真皮淋巴管内皮细胞增生,甚至使管腔完全闭塞,皮下淋巴管壁可有明显肌层肥厚。发病部位最多见于下肢、阴囊、女阴等处,其次为手臂及乳房。以下肢的象皮肿最多见,约占90%,常为双侧性,由踝部和足背部开始,逐渐扩展到小腿甚至大腿,下肢可比正常增粗2~3倍(图19-17)。阴囊象皮肿大小不等,小者如拳头大,大者可大如篮球或更大,甚至可下垂到膝部以下。

图19-17丝虫病晚期的下肢象皮肿
关于象皮肿的发生机制,过去一般认为系由于淋巴管的慢性机械性阻塞而引起,即淋巴液由于回流障碍而蓄积在皮肤及皮下组织,因淋巴液的蛋白质含量高,刺激纤维组织大量增生,使局部皮肤增厚、变硬,而局部的反复链球菌低度感染,则促进了象皮肿的发生和发展。但近年来通过淋巴系统造影术发现,象皮肿患者的淋巴系统大多并不见有阻塞,因而认为不是淋巴流的机械性闭塞而是由于淋巴循环严重的病理生理动力学改变所致的淋巴循环障碍引起象皮肿。也有认为象皮肿的局部反应属于Arthus变态反应。
象皮肿的发展很慢,一般都在感染后的10~15年以上才能达到显著程度。患者的血液中大多已找不到微丝蚴,可能因成虫已死亡,不能产生微丝蚴,或因淋巴循环障碍,微丝蚴不能进入血流之故。
第六节 包虫病
包虫病(hydatid disease)是人类感染棘球绦虫的幼虫所致的疾病,故又称棘球蚴病。本病几乎遍布全世界,在我国甘肃、宁夏、青海、新疆、陕西、内蒙及四川西部等畜牧地区为常见的寄生虫病。在人类绦虫病中,本病的危害最为严重。
寄生于人体的幼虫有细粒棘绦虫(Echincoccus granulosus)及泡状(或多房)棘球绦虫(Echinococcus alveolaris s.multilocularis)两种,引起细粒棘球蚴病和泡状棘球蚴病。在我国,以前者更为常见。
一、细粒棘球蚴病
【病因及感染途径】
细粒棘球绦虫是绦虫类最细小的一种,其成虫主要寄生在狗的小肠内,狗是最重要的终宿主和传染源,亦寄生于狼等其他肉食动物。细粒棘球绦虫的虫体长2~7mm,雌雄同体,由一个头节和三个体节(即幼节、成节和孕节)组成。头节顶有顶突及大小两圈小钩,并有四个吸盘。孕节内含有感染性的虫卵。被孕节或虫卵污染的水或食物中间宿主如羊、牛、猪、家兔、骆驼等家畜及人食入后,即在胃或十二指肠内孵化,六钩蚴脱壳而出,先附着于小肠粘膜,再钻入肠壁血管,随血流经门静脉到达肝,故肝包虫病最多见。少部分可通过肝经右心到肺,极少数可通过肺循环而到达全身其他器官。但六钩蚴也可从肠壁侵入淋巴管,经胸导管直接进入血流而至全身各处。幼虫经过数月的发育,即成为囊状幼虫,称为棘球蚴或包虫囊。棘球蚴内含有很多原头蚴(头节),如果含棘球蚴的器官被狗、狼等吞食,其中的每一个原头蚴均能在小肠壁发育为成虫。以后陆续排出孕节和虫卵,造成污染和感染。
【病理变化】
棘球蚴主要引起肝病变(占70%),其次为肺(占20%~30%)。肌肉、心、脾、肾、脑、骨、眼眶等更少见(10%)。近年来,肌肉的感染有增多趋势(1.4%~5.4%)。
六钩蚴侵入组织后,可引起周围组织巨噬细胞和嗜酸性粒细胞浸润。大多数六钩蚴会死去,仅少数存活发育成包虫囊。包虫囊生长极为缓慢,感染5个月后直径仅达1cm左右,约经5~20年可达到巨大程度,最大者可达50cm。包虫囊周围有类上皮细胞、异物巨细胞、嗜酸性粒细胞浸润及纤维母细胞增生,最终可形成纤维性包膜,也称为外囊。外囊的厚薄与囊肿形成的时间有关,一般约3~5mm,也可达1cm左右。
包虫囊壁分为内、外两层,内层为生发层,厚约22~25μm,由单层或多层的生发细胞构成,具有显著的繁殖能力。生发层细胞向内芽生,可在囊内壁形成无数小突起,渐变成单层小囊泡,即生发囊(图19-18)。生发囊脱落,即变子囊,其内壁又可生出5~30个原头蚴。子囊结构与母囊相同,还可再产生生发囊或孙囊。在较老的包虫囊内,子囊可多达数百个。生发层偶也向外芽生形成外生囊。
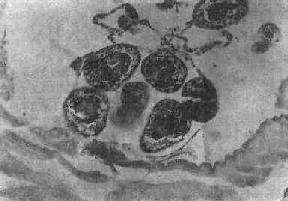
图19-18 包虫囊肿
囊壁外层为角皮层,内为生发层,并有生发囊形成,其内可见多数原头蚴
包虫囊壁的外层为角皮层,呈白色半透明状,如粉皮,厚约3~4mm,具有吸收营养物质及保护生发层作用。镜下为红染平行的板层状结构。包虫囊内含无色或微黄色液体,液量由数百到数千毫升,甚至可多达2万毫升。囊液中所含的蛋白质具有抗原性。囊壁破裂后可引起周围组织发生局部过敏性反应,严重者可发生过敏性休克。如果子囊破裂,大量头节混入囊内液体,在其中自由游动,即形成棘球蚴砂(或包虫砂)。棘球蚴可生存达40年之久甚至更长,但可因损伤、感染而退化死亡,此时母囊及子囊发生钙化,囊内液化被吸收浓缩变为胶泥样物,其中仍可见原头蚴。
【主要器官病变及其后果】
1.肝包虫囊肿 为包虫病中最常见者,多见于右叶,囊肿多为单个,位于膈面,向腹腔突出,也可为多个。
肝包虫囊肿生长缓慢,逐渐增大可致周围肝细胞压迫性萎缩或变性,其外纤维组织增生,形成一层纤维性外囊。肝内小胆管及血管也常因受压而移位,或被包入囊壁内。临床上巨大包虫囊大都位于肝,引起肝区肿大,待囊肿退化变性后,则囊肿随之变小。
肝包虫囊肿长大后其主要而常见的并发症为继发感染和囊肿破裂。继发感染主要由于被包入外囊中的小胆管破入包虫囊肿腔内引起,也可因外伤、穿刺及血道感染引起。感染后引起的病理变化似肝脓肿,但症状较轻。肝包虫囊肿破裂为常见且严重的并发症,多由继发感染、外伤或穿刺引起,以破入腹腔的后果最为严重,此时可导致过敏性休克而致患者死亡,还产生腹腔内继发性包虫囊肿。如子囊破入胆管或肝静脉内,可造成胆道阻塞及肺动脉栓塞。
2.肺包虫囊肿 肺包虫囊肿多见于右肺和下叶,通常为单个,多发者少见。囊肿多位于肺的周边区。由于肺组织疏松和血循丰富及胸腔负压吸引等影响,故肺包虫囊肿生长较快,可压迫周围肺组织,引起肺萎陷和纤维化。由于肺包虫囊肿的纤维外膜及包虫囊的角皮层较薄,故易破裂。破入支气管,则囊内容物和囊可被咳出而自行痊愈;大量囊液破入支气管时可引起窒息。少数病例可破入胸腔,引起包虫性胸膜炎。
二、泡状棘球蚴病
泡状棘球蚴病比较少见,主要侵犯肝。在我国新疆、青海、四川、甘肃、内蒙等地有病例报告。
【病因、感染途径】
泡状棘球绦虫的成虫与细粒棘球绦虫相似,但虫体较短(1.4~3.4mm),体节2~5节,偶为6节,吸盘及大小钩均较短小,头钩和睾丸数亦较少。泡球蚴不形成大囊泡,而成海绵状。囊泡生长较快;子囊为外生性,原头蚴数少,以上均与细粒棘球蚴不同。
泡状棘球绦虫的成虫主要寄生于孤,其次为狗、狼、猫等。中间宿主主要为鼠类,人类也可被虫卵感染,但并非适宜的中间宿主。
病变绝大多数泡状棘球囊肿见于肝,一般呈单个巨块型,有时为结节型,或两者兼有。泡球蚴囊泡常呈灰白色,质较硬,由无数小囊泡集合而成海绵状,与周围组织分界不清(图19-19)。囊泡内容物为豆腐渣样蚴体碎屑和小泡。陈旧病灶的中央因营养不佳常发生变性、坏死,或溶解呈胶冻状液体。如继发感染,可酷似脓肿。泡状囊肿外周无纤维包膜,向外芽生性子囊可以像癌肿一样向周围组织浸润,并可侵入血管或淋巴管,转移到肺、脑、脾、肾、肾上腺及心脏等处,甚至偶然可见于肝门淋巴结内,因此肉眼上易误诊为肝癌。镜下,在肝组织中散在大小不等的泡状蚴小囊泡,一般仅见角皮层,偶尔有单细胞性生发层,偶见原头蚴。囊泡周围有嗜酸性粒细胞浸润,伴有结核样肉芽组织形成及纤维组织增生。囊泡间的肝组织常发生凝固性坏死(图19-20)。病变周围肝组织常有肝细胞萎缩、变性或坏死及淤胆现象。最后可导致肝硬变、黄疸、门静脉高压和肝功能衰竭及恶病质。

图19-19 肝泡状棘球蚴病(巨块型)
切面上见由无数小囊泡集合而成
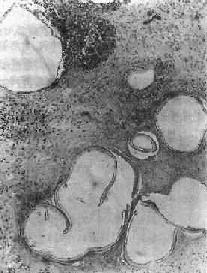
图19-20 肝泡状棘球蚴病
可见多数大小不等的小囊泡,仅见角皮层而不见生发层,囊内无头节,囊泡间组织呈凝固性坏死
肺泡球蚴病少见,多数由肝经血流迁徙而来,或由肝直接蔓延而来。泡状蚴穿出血管后,先在细支气管寄生,引起增生性炎及血管内膜炎。病变也可呈肿块状,类似肿瘤。